当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electrostatics does not dictate the slip-stacked arrangement of aromatic π–π interactions
Chemical Science ( IF 7.6 ) Pub Date : 2020-06-05 , DOI: 10.1039/d0sc02667k Kevin Carter-Fenk 1, 2, 3, 4 , John M. Herbert 1, 2, 3, 4
Chemical Science ( IF 7.6 ) Pub Date : 2020-06-05 , DOI: 10.1039/d0sc02667k Kevin Carter-Fenk 1, 2, 3, 4 , John M. Herbert 1, 2, 3, 4
Affiliation
|
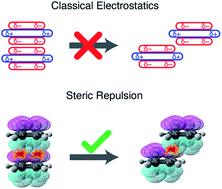
Benzene dimer has long been an archetype for π-stacking. According to the Hunter–Sanders model, quadrupolar electrostatics favors an edge-to-face CH⋯π geometry but competes with London dispersion that favors cofacial π-stacking, with a compromise “slip-stacked” structure emerging as the minimum-energy geometry. This model is based on classical electrostatics, however, and neglects charge penetration. A fully quantum-mechanical analysis, presented here, demonstrates that electrostatics actually exerts very little influence on the conformational landscape of (C6H6)2. Electrostatics also cannot explain the slip-stacked arrangement of C6H6⋯C6F6, where the sign of the quadrupolar interaction is reversed. Instead, the slip-stacked geometry emerges in both systems due to competition between dispersion and Pauli repulsion, with electrostatics as an ambivalent spectator. This revised interpretation helps to rationalize the persistence of offset π-stacking in larger polycyclic aromatic hydrocarbons and across the highly varied electrostatic environments that characterize π–π interactions in proteins.
中文翻译:

静电不会决定芳族π-π相互作用的滑动堆积排列
苯二聚体一直是π堆积的原型。根据Hunter-Sanders模型,四极静电学倾向于边对面的CH⋯π几何形状,但是与伦敦分散相竞争,后者有利于界面π堆叠,而折衷的“滑移堆叠”结构则成为最小能量几何。但是,该模型基于经典静电学,忽略了电荷渗透。此处提供的完全量子力学分析表明,静电实际上对(C 6 H 6)2的构象态几乎没有影响。静电学也无法解释C 6 H 6 ⋯C 6 F 6的粉滑堆积排列,其中四极相互作用的符号相反。取而代之的是,由于色散和保利斥力之间的竞争,在两个系统中都出现了滑动堆叠的几何形状,其中静电是矛盾的旁观者。修改后的解释有助于合理化较大的多环芳烃中以及在表征蛋白质中π-π相互作用的高度变化的静电环境中存在的偏移π堆积的持久性。
更新日期:2020-07-08
中文翻译:
静电不会决定芳族π-π相互作用的滑动堆积排列
苯二聚体一直是π堆积的原型。根据Hunter-Sanders模型,四极静电学倾向于边对面的CH⋯π几何形状,但是与伦敦分散相竞争,后者有利于界面π堆叠,而折衷的“滑移堆叠”结构则成为最小能量几何。但是,该模型基于经典静电学,忽略了电荷渗透。此处提供的完全量子力学分析表明,静电实际上对(C 6 H 6)2的构象态几乎没有影响。静电学也无法解释C 6 H 6 ⋯C 6 F 6的粉滑堆积排列,其中四极相互作用的符号相反。取而代之的是,由于色散和保利斥力之间的竞争,在两个系统中都出现了滑动堆叠的几何形状,其中静电是矛盾的旁观者。修改后的解释有助于合理化较大的多环芳烃中以及在表征蛋白质中π-π相互作用的高度变化的静电环境中存在的偏移π堆积的持久性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号