当前位置:
X-MOL 学术
›
Magn. Reson. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Local atomic and electronic structure in the LiVPO4 (F,O) Tavorite-type materials from Solid State NMR combined with DFT calculations
Magnetic Resonance in Chemistry ( IF 1.9 ) Pub Date : 2020-07-01 , DOI: 10.1002/mrc.5059 Tahya Bamine 1, 2 , Edouard Boivin 1, 2, 3 , Christian Masquelier 2, 3, 4 , Laurence Croguennec 1, 2, 4 , Elodie Salager 2, 5 , Dany Carlier 1, 2, 4
Magnetic Resonance in Chemistry ( IF 1.9 ) Pub Date : 2020-07-01 , DOI: 10.1002/mrc.5059 Tahya Bamine 1, 2 , Edouard Boivin 1, 2, 3 , Christian Masquelier 2, 3, 4 , Laurence Croguennec 1, 2, 4 , Elodie Salager 2, 5 , Dany Carlier 1, 2, 4
Affiliation
|
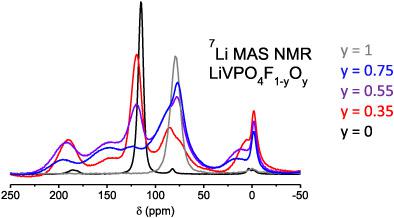
7 Li, 31 P and 19 F solid state NMR spectroscopy was used to investigate the local arrangement of Oxygen and Fluorine in LiVPO4 F1-y Oy materials, interesting as positive electrode materials for Li-ion batteries. From the evolution of the 1D spectra versus y, 2D 7 Li RFDR experiments combined and a tentative signal assignment based on DFT calculations, it appears that F and O are not randomly dispersed on the bridging X position between two X-VO4 -X octahedra (X = O or F), but tend to segregate at a local scale. Using DFT calculations, we analyzed the impact of the different local environments on the local electronic structure. Depending on the nature of the VO4 X2 environments, vanadium ions are either in the +III or in the +IV oxidation state and can exhibit different distributions of their unpaired electron(s) on the d orbitals. Based on those different local electronic structures and on the computed Fermi contact shifts, we discuss the impact on the spin transfer mechanism on adjacent nuclei and propose tentative signal assignments. The O/F clustering tendency is discussed in relation with the formation of short VIV =O vanadyl bonds with a very specific electronic structure and possible cooperative effect along the chain.
中文翻译:

来自固态核磁共振的 LiVPO4 (F,O) Tavorite 型材料中的局部原子和电子结构结合 DFT 计算
7 Li、 31 P 和 19 F 固态核磁共振光谱用于研究氧和氟在 LiVPO4 F1-y Oy 材料中的局部排列,该材料可用作锂离子电池的正极材料。从一维光谱与 y、二维 7 Li RFDR 实验相结合的演变以及基于 DFT 计算的初步信号分配来看,F 和 O 似乎不是随机分散在两个 X-VO4 -X 八面体之间的桥接 X 位置上( X = O 或 F),但倾向于在局部范围内分离。使用 DFT 计算,我们分析了不同局部环境对局部电子结构的影响。根据 VO4 X2 环境的性质,钒离子处于 +III 或 +IV 氧化态,并且可以在 d 轨道上表现出不同的未配对电子分布。基于这些不同的局部电子结构和计算出的费米接触位移,我们讨论了对相邻原子核的自旋转移机制的影响,并提出了初步的信号分配。讨论了 O/F 聚集趋势与短 VIV =O 氧钒键的形成有关,该键具有非常特定的电子结构和沿链可能的协同效应。
更新日期:2020-07-01
中文翻译:
来自固态核磁共振的 LiVPO4 (F,O) Tavorite 型材料中的局部原子和电子结构结合 DFT 计算
7 Li、 31 P 和 19 F 固态核磁共振光谱用于研究氧和氟在 LiVPO4 F1-y Oy 材料中的局部排列,该材料可用作锂离子电池的正极材料。从一维光谱与 y、二维 7 Li RFDR 实验相结合的演变以及基于 DFT 计算的初步信号分配来看,F 和 O 似乎不是随机分散在两个 X-VO4 -X 八面体之间的桥接 X 位置上( X = O 或 F),但倾向于在局部范围内分离。使用 DFT 计算,我们分析了不同局部环境对局部电子结构的影响。根据 VO4 X2 环境的性质,钒离子处于 +III 或 +IV 氧化态,并且可以在 d 轨道上表现出不同的未配对电子分布。基于这些不同的局部电子结构和计算出的费米接触位移,我们讨论了对相邻原子核的自旋转移机制的影响,并提出了初步的信号分配。讨论了 O/F 聚集趋势与短 VIV =O 氧钒键的形成有关,该键具有非常特定的电子结构和沿链可能的协同效应。










































 京公网安备 11010802027423号
京公网安备 11010802027423号