当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
PyCDFT : A Python package for constrained density functional theory
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-06-04 , DOI: 10.1002/jcc.26354 He Ma 1, 2 , Wennie Wang 3 , Siyoung Kim 3 , Man-Hin Cheng 3 , Marco Govoni 2, 3 , Giulia Galli 1, 2, 3
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-06-04 , DOI: 10.1002/jcc.26354 He Ma 1, 2 , Wennie Wang 3 , Siyoung Kim 3 , Man-Hin Cheng 3 , Marco Govoni 2, 3 , Giulia Galli 1, 2, 3
Affiliation
|
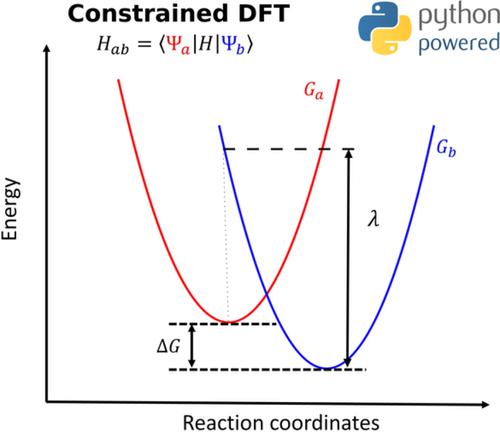
We present PyCDFT, a Python package to compute diabatic states using constrained density functional theory (CDFT). PyCDFT provides an object‐oriented, customizable implementation of CDFT, and allows for both single‐point self‐consistent‐field calculations and geometry optimizations. PyCDFT is designed to interface with existing density functional theory (DFT) codes to perform CDFT calculations where constraint potentials are added to the Kohn–Sham Hamiltonian. Here, we demonstrate the use of PyCDFT by performing calculations with a massively parallel first‐principles molecular dynamics code, Qbox, and we benchmark its accuracy by computing the electronic coupling between diabatic states for a set of organic molecules. We show that PyCDFT yields results in agreement with existing implementations and is a robust and flexible package for performing CDFT calculations. The program is available at https://dx.doi.org/10.5281/zenodo.3821097.
中文翻译:

PyCDFT:约束密度泛函理论的 Python 包
我们提出了 PyCDFT,这是一个使用约束密度泛函理论 (CDFT) 计算非绝热状态的 Python 包。PyCDFT 提供了面向对象的、可定制的 CDFT 实现,并允许单点自洽场计算和几何优化。PyCDFT 旨在与现有的密度泛函理论 (DFT) 代码接口,以执行 CDFT 计算,其中将约束势添加到 Kohn-Sham 哈密顿量。在这里,我们通过使用大规模并行第一性原理分子动力学代码 Qbox 执行计算来演示 PyCDFT 的使用,并通过计算一组有机分子的非绝热状态之间的电子耦合来衡量其准确性。我们表明 PyCDFT 产生的结果与现有实现一致,并且是用于执行 CDFT 计算的强大而灵活的包。该程序可在 https://dx.doi.org/10.5281/zenodo.3821097 上获得。
更新日期:2020-06-04
中文翻译:
PyCDFT:约束密度泛函理论的 Python 包
我们提出了 PyCDFT,这是一个使用约束密度泛函理论 (CDFT) 计算非绝热状态的 Python 包。PyCDFT 提供了面向对象的、可定制的 CDFT 实现,并允许单点自洽场计算和几何优化。PyCDFT 旨在与现有的密度泛函理论 (DFT) 代码接口,以执行 CDFT 计算,其中将约束势添加到 Kohn-Sham 哈密顿量。在这里,我们通过使用大规模并行第一性原理分子动力学代码 Qbox 执行计算来演示 PyCDFT 的使用,并通过计算一组有机分子的非绝热状态之间的电子耦合来衡量其准确性。我们表明 PyCDFT 产生的结果与现有实现一致,并且是用于执行 CDFT 计算的强大而灵活的包。该程序可在 https://dx.doi.org/10.5281/zenodo.3821097 上获得。










































 京公网安备 11010802027423号
京公网安备 11010802027423号