当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Hydrogen Bonding of Ammonia with (H,OH)-Si(001) Revealed by Experimental and Ab Initio Photoelectron Spectroscopy.
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2020-06-03 , DOI: 10.1021/acs.jpca.0c03458 Lucía Pérez Ramírez 1 , Jean-Jacques Gallet 1, 2 , Fabrice Bournel 1, 2 , Florence Lim 1 , Stéphane Carniato 1 , François Rochet 1 , Oleg V Yazyev 3 , Alfredo Pasquarello 3 , Elena Magnano 4, 5 , Federica Bondino 4
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2020-06-03 , DOI: 10.1021/acs.jpca.0c03458 Lucía Pérez Ramírez 1 , Jean-Jacques Gallet 1, 2 , Fabrice Bournel 1, 2 , Florence Lim 1 , Stéphane Carniato 1 , François Rochet 1 , Oleg V Yazyev 3 , Alfredo Pasquarello 3 , Elena Magnano 4, 5 , Federica Bondino 4
Affiliation
|
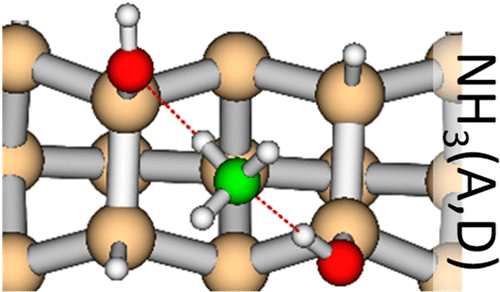
Combining experimental and ab initio core-level photoelectron spectroscopy (periodic DFT and quantum chemistry calculations), we elucidated how ammonia molecules bond to the hydroxyls of the (H,OH)-Si(001) model surface at a temperature of 130 K. Indeed, theory evaluated the magnitude and direction of the N 1s (and O 1s) chemical shifts according to the nature (acceptor or donor) of the hydrogen bond and, when confronted to experiment, showed unambiguously that the probe molecule makes one acceptor and one donor bond with a pair of hydroxyls. The consistency of our approach was proved by the fact that the identified adsorption geometries are precisely those that have the largest binding strength to the surface, as calculated by periodic DFT. Real-time core-level photoemission enabled measurement of the adsorption kinetics of H-bonded ammonia and its maximum coverage (0.37 ML) under 1.5 × 10–9 mbar. Experimental desorption free energies were compared to the magnitude of the adsorption energies provided by periodic DFT calculations. Minority species were also detected on the surface. As in the case of H-bonded ammonia, DFT core-level calculations were instrumental to attribute these minority species to datively bonded ammonia molecules, associated with isolated dangling bonds remaining on the surface, and to dissociated ammonia molecules, resulting largely from beam damage.
中文翻译:

实验和从头算光电子能谱揭示了氨与(H,OH)-Si(001)的氢键。
结合实验和从头开始的核能级光电子能谱(周期性DFT和量子化学计算),我们阐明了在130 K的温度下氨分子如何与(H,OH)-Si(001)模型表面的羟基键合。 ,理论根据氢键的性质(受体或供体)评估了N 1s(和O 1s)化学位移的大小和方向,并且在面对实验时明确表明探针分子是一个受体和一个供体与一对羟基键合。我们的方法的一致性由以下事实证明:经定期DFT计算,所确定的吸附几何形状恰好是对表面具有最大结合强度的那些几何形状。–9毫巴。将实验的解吸自由能与定期DFT计算提供的吸附能的大小进行了比较。在表面上也检测到少数物种。与氢键结合的氨的情况一样,DFT核心水平计算有助于将这些少数物种归因于键合的氨分子(与表面上残留的孤立的悬空键相关)以及离解的氨分子,这主要是由束损伤造成的。
更新日期:2020-07-02
中文翻译:
实验和从头算光电子能谱揭示了氨与(H,OH)-Si(001)的氢键。
结合实验和从头开始的核能级光电子能谱(周期性DFT和量子化学计算),我们阐明了在130 K的温度下氨分子如何与(H,OH)-Si(001)模型表面的羟基键合。 ,理论根据氢键的性质(受体或供体)评估了N 1s(和O 1s)化学位移的大小和方向,并且在面对实验时明确表明探针分子是一个受体和一个供体与一对羟基键合。我们的方法的一致性由以下事实证明:经定期DFT计算,所确定的吸附几何形状恰好是对表面具有最大结合强度的那些几何形状。–9毫巴。将实验的解吸自由能与定期DFT计算提供的吸附能的大小进行了比较。在表面上也检测到少数物种。与氢键结合的氨的情况一样,DFT核心水平计算有助于将这些少数物种归因于键合的氨分子(与表面上残留的孤立的悬空键相关)以及离解的氨分子,这主要是由束损伤造成的。


























 京公网安备 11010802027423号
京公网安备 11010802027423号