当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effect of Protein Flexibility from Coarse-Grained Elastic Network Parameterizations on the Calculation of Free Energy Profiles of Ligand Binding.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-06-04 , DOI: 10.1021/acs.jctc.0c00418 Hugo A L Filipe 1 , Margarida I M Esteves 1 , César A Henriques 2 , Filipe E Antunes 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-06-04 , DOI: 10.1021/acs.jctc.0c00418 Hugo A L Filipe 1 , Margarida I M Esteves 1 , César A Henriques 2 , Filipe E Antunes 1
Affiliation
|
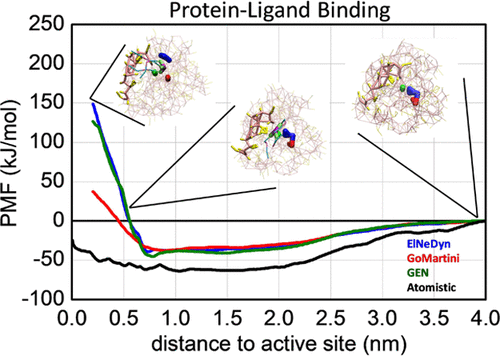
The characterization of the affinity and binding mechanism of specific molecules to a protein active site is scientifically and industrially relevant for many applications. In principle, this information can be obtained using molecular dynamics (MD) simulations by calculating the free energy profile of the process. However, this is a computationally demanding calculation. Currently, coarse-grained (CG) force fields are very well implemented for MD simulations of biomolecular systems. These computationally efficient force fields are a major advantage to the study of large model systems and/or those requiring long simulation times. The Martini model is currently one of the most popular CG force fields for these systems. For the specific case of protein simulations, to correctly maintain the macromolecular three-dimensional structure, the Martini model needs to include an elastic network (EN). In this work, the effect of protein flexibility, as induced by three EN models compatible with the Martini force field, was tested on the calculation of free energy profiles for protein-ligand binding. The EN models used were ElNeDyn, GoMartini, and GEN. The binding of triolein (TOG) and triacetin (TAG) to a lipase protein (thermomyces lanuginosa lipase-TLL) was used as a case study. The results show that inclusion of greater flexibility in the CG parameterization of proteins is of high importance in the calculation of the free energy profiles of protein-ligand systems. However, care must be taken in order to avoid unjustified large protein deformations. In addition, due to molecular flexibility there may be no absolute need for the center of the ligand to reach the center of the protein-binding site. The calculation of the energy profile to a distance of about 0.5 nm from the active site center can be sufficient to differentiate the affinity of different ligands to a protein.
中文翻译:

粗粒弹性网络参数化对蛋白质柔韧性的影响,对配体结合自由能分布的计算。
特定分子对蛋白质活性位点的亲和力和结合机制的表征在科学和工业上与许多应用有关。原则上,可以使用分子动力学(MD)模拟通过计算过程的自由能曲线来获取此信息。但是,这是一个计算要求很高的计算。当前,对于生物分子系统的MD模拟,很好地实现了粗粒度(CG)力场。这些计算有效的力场是研究大型模型系统和/或需要较长仿真时间的系统的主要优势。目前,马提尼模型是这些系统最受欢迎的CG力场之一。对于蛋白质模拟的特定情况,要正确维护大分子的三维结构,Martini模型需要包含一个弹性网络(EN)。在这项工作中,测试了由三种与马提尼力场兼容的EN模型诱导的蛋白质柔韧性的影响,以计算蛋白质与配体结合的自由能。使用的EN模型是ElNeDyn,GoMartini和GEN。三油精(TOG)和三醋精(TAG)与脂肪酶蛋白(嗜热霉菌脂肪酶(TLL)用作案例研究。结果表明,在蛋白质的CG参数设置中包含更大的灵活性在计算蛋白质-配体系统的自由能分布中非常重要。但是,必须注意避免不合理的大蛋白变形。另外,由于分子的柔性,可能绝对不需要配体的中心到达蛋白质结合位点的中心。距活性位点中心约0.5 nm处的能量分布图计算足以区分不同配体对蛋白质的亲和力。
更新日期:2020-07-14
中文翻译:
粗粒弹性网络参数化对蛋白质柔韧性的影响,对配体结合自由能分布的计算。
特定分子对蛋白质活性位点的亲和力和结合机制的表征在科学和工业上与许多应用有关。原则上,可以使用分子动力学(MD)模拟通过计算过程的自由能曲线来获取此信息。但是,这是一个计算要求很高的计算。当前,对于生物分子系统的MD模拟,很好地实现了粗粒度(CG)力场。这些计算有效的力场是研究大型模型系统和/或需要较长仿真时间的系统的主要优势。目前,马提尼模型是这些系统最受欢迎的CG力场之一。对于蛋白质模拟的特定情况,要正确维护大分子的三维结构,Martini模型需要包含一个弹性网络(EN)。在这项工作中,测试了由三种与马提尼力场兼容的EN模型诱导的蛋白质柔韧性的影响,以计算蛋白质与配体结合的自由能。使用的EN模型是ElNeDyn,GoMartini和GEN。三油精(TOG)和三醋精(TAG)与脂肪酶蛋白(嗜热霉菌脂肪酶(TLL)用作案例研究。结果表明,在蛋白质的CG参数设置中包含更大的灵活性在计算蛋白质-配体系统的自由能分布中非常重要。但是,必须注意避免不合理的大蛋白变形。另外,由于分子的柔性,可能绝对不需要配体的中心到达蛋白质结合位点的中心。距活性位点中心约0.5 nm处的能量分布图计算足以区分不同配体对蛋白质的亲和力。








































 京公网安备 11010802027423号
京公网安备 11010802027423号