Computational and Theoretical Chemistry ( IF 3.0 ) Pub Date : 2020-06-03 , DOI: 10.1016/j.comptc.2020.112890 H. Khettal , M.F. Haroun , M. Boukelkoul
|
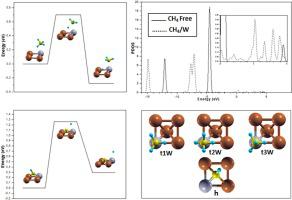
First principle density functional theory calculations (DFT GGA-PW91) have been employed to study the activity of W-doped Cu(1 0 0) surface for methane dissociation. We studied methane site preference for CH4, CH3, CH2, CH, C and H species at a coverage of a 0.25 monolayer. From the calculated adsorption energies, we found that W-doping stabilizes better all species. The top tungsten site is the most stable for methane, methyl and hydrogen. However, the methyn and carbon atoms prefer the fourfold hollow site, while methylene prefer bridge site. In addition, the electronic structure analysis of methane and surface showed a weak chemical interaction. Furthermore, methane complete dehydrogenation mechanism have been investigated in terms of the thermodynamics and kinetics. The results revealed that the W-Cu(1 0 0) is more reactive surface for methane dissociation, thermodynamically and kinetically, rather than others Cu-based catalysts.
中文翻译:
CH 4在W-Cu(1 0 0)表面吸附和解离的理论研究
第一原理密度泛函理论计算(DFT GGA-PW91)已用于研究W掺杂的Cu(1 0 0)表面对甲烷的离解活性。我们研究了CH 4,CH 3, CH 2的甲烷位点偏好,CH,C和H物种,覆盖度为0.25单层。根据计算出的吸附能,我们发现W掺杂能更好地稳定所有物种。顶部钨位置对于甲烷,甲基和氢最稳定。然而,次甲基和碳原子更喜欢四重空心位点,而亚甲基更喜欢桥位。此外,甲烷和表面的电子结构分析显示出较弱的化学相互作用。此外,已经从热力学和动力学方面研究了甲烷完全脱氢机理。结果表明,W-Cu(1 0 0)比其它基于铜的催化剂在热力学和动力学上对甲烷的离解反应性更高。









































 京公网安备 11010802027423号
京公网安备 11010802027423号