当前位置:
X-MOL 学术
›
Phys. Status Solidi B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Dynamics Study of Thermal Conductivity of Silver Chalcogenides
Physica Status Solidi (B) - Basic Solid State Physics ( IF 1.5 ) Pub Date : 2020-06-03 , DOI: 10.1002/pssb.202000183 Shogo Fukushima 1 , Kohei Shimamura 1 , Akihide Koura 1 , Fuyuki Shimojo 1
Physica Status Solidi (B) - Basic Solid State Physics ( IF 1.5 ) Pub Date : 2020-06-03 , DOI: 10.1002/pssb.202000183 Shogo Fukushima 1 , Kohei Shimamura 1 , Akihide Koura 1 , Fuyuki Shimojo 1
Affiliation
|
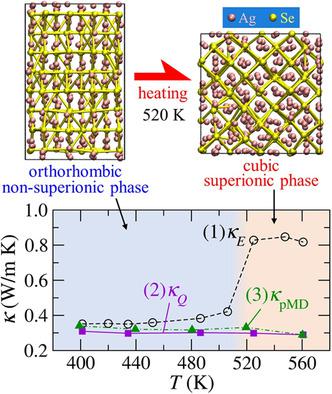
To obtain an accurate thermal conductivity of Ag2Se considering heat‐flux effects, computational requirements such as simulation times and number of atoms are investigated for two methods: the Green–Kubo (GK) formula in equilibrium molecular dynamics (EMD) simulations and perturbed MD (pMD). An empirical interatomic potential is used, which reproduces both the superionic and nonsuperionic phases of Ag2Se. It is found that the accurate thermal conductivity based on the GK formula requires at least the correlation time of 12 ps, the simulation time of 24 ns, and the number of atoms of 3000. In contrast, for the pMD, the simulation time of ≈1.2 ns is needed to obtain a comparable thermal conductivity. It is also confirmed that the heat flux must be considered in MD simulations to qualitatively reproduce the temperature dependence of thermal conductivity observed experimentally.
中文翻译:

银硫属元素化物导热系数的分子动力学研究
为了获得考虑到热通量效应的准确的Ag 2 Se导热系数,针对两种方法研究了仿真时间和原子数等计算要求:平衡分子动力学(EMD)仿真中的Green-Kubo(GK)公式,并对其进行了扰动MD(pMD)。使用经验原子间势,该势可重现Ag 2的超离子相和非超离子相硒 发现基于GK公式的准确热导率至少需要12 ps的相关时间,24 ns的仿真时间和3000个原子数。相反,对于pMD,仿真时间为要获得可比的导热率,需要1.2 ns。还证实在MD模拟中必须考虑热通量,以定性地再现实验观察到的热导率的温度依赖性。
更新日期:2020-06-03
中文翻译:
银硫属元素化物导热系数的分子动力学研究
为了获得考虑到热通量效应的准确的Ag 2 Se导热系数,针对两种方法研究了仿真时间和原子数等计算要求:平衡分子动力学(EMD)仿真中的Green-Kubo(GK)公式,并对其进行了扰动MD(pMD)。使用经验原子间势,该势可重现Ag 2的超离子相和非超离子相硒 发现基于GK公式的准确热导率至少需要12 ps的相关时间,24 ns的仿真时间和3000个原子数。相反,对于pMD,仿真时间为要获得可比的导热率,需要1.2 ns。还证实在MD模拟中必须考虑热通量,以定性地再现实验观察到的热导率的温度依赖性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号