当前位置:
X-MOL 学术
›
Isr. J. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Evaluation of Polaron Transport in Solids from First‐principles
Israel Journal of Chemistry ( IF 2.3 ) Pub Date : 2020-01-24 , DOI: 10.1002/ijch.201900101 Yuriy Natanzon 1 , Amram Azulay 1 , Yaron Amouyal 1
Israel Journal of Chemistry ( IF 2.3 ) Pub Date : 2020-01-24 , DOI: 10.1002/ijch.201900101 Yuriy Natanzon 1 , Amram Azulay 1 , Yaron Amouyal 1
Affiliation
|
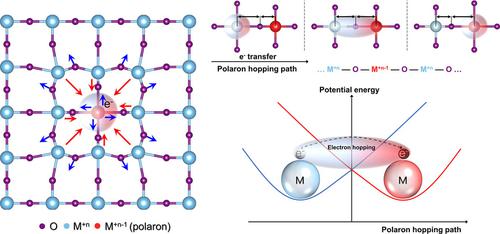
Polarons are formed in polar or ionic solids, either molecular or crystalline, due to local distortions of the lattice induced by charge carriers. Polaron hopping is the primary mechanism of charge transport in these materials, such as functional ceramic compounds, with applications in photovoltaics, thermoelectrics, two‐dimensional electron gas transistors, magnetic sensors, spin valve devices, and memories. Understanding the fundamental physics of polaron hopping is, therefore, of prime technological importance. This article provides a brief physical background of polarons and their hopping mechanism, focusing on first‐principles calculations of polaron properties. Herein, we review recent selected studies applying the density functional theory (DFT), and describe the merits and challenges in applying DFT for such calculations, highlighting the need to address both electronic and vibrational aspects. The vibrational component of the polaron is evaluated based on structural and total energy calculations, whereas the electronic component is derived from both total energy and electron density calculations. To address the most compelling challenge of calculating polaron properties using DFT, which is the issue of electron localization, we propose to employ calculations of selected vibrational properties, such as the sound velocity, shear modulus, and Grüneisen parameter, to represent the polaron hopping energy; all of which originate from the stiffness of inter‐atomic bonds. Such methodology is expected to be more straightforward than the existing ones, however demands standardization.
中文翻译:

第一性原理对固体中极化子迁移的评估
由于电荷载流子引起的晶格局部变形,极化子以分子或晶体的极性或离子固体形式形成。极化子跳跃是这些材料(例如功能性陶瓷化合物)中电荷传输的主要机制,应用于光伏,热电,二维电子气晶体管,磁性传感器,自旋阀设备和存储器。因此,了解极化子跳跃的基本物理原理至关重要。本文简要介绍了极化子及其跳变机理的物理背景,重点介绍极化子特性的第一性原理计算。在此,我们回顾了最近应用密度泛函理论(DFT)进行的一些研究,并描述了将DFT用于此类计算的优缺点。强调需要解决电子和振动方面的问题。极化子的振动分量是根据结构和总能量计算进行评估的,而电子分量是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。极化子的振动分量是根据结构和总能量计算进行评估的,而电子分量是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。极化子的振动分量是根据结构和总能量计算进行评估的,而电子分量是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。而电子组件是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。而电子组件是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。和Grüneisen参数,代表极化子跳跃能量;所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。和Grüneisen参数,代表极化子跳跃能量;所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。
更新日期:2020-01-24
中文翻译:
第一性原理对固体中极化子迁移的评估
由于电荷载流子引起的晶格局部变形,极化子以分子或晶体的极性或离子固体形式形成。极化子跳跃是这些材料(例如功能性陶瓷化合物)中电荷传输的主要机制,应用于光伏,热电,二维电子气晶体管,磁性传感器,自旋阀设备和存储器。因此,了解极化子跳跃的基本物理原理至关重要。本文简要介绍了极化子及其跳变机理的物理背景,重点介绍极化子特性的第一性原理计算。在此,我们回顾了最近应用密度泛函理论(DFT)进行的一些研究,并描述了将DFT用于此类计算的优缺点。强调需要解决电子和振动方面的问题。极化子的振动分量是根据结构和总能量计算进行评估的,而电子分量是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。极化子的振动分量是根据结构和总能量计算进行评估的,而电子分量是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。极化子的振动分量是根据结构和总能量计算进行评估的,而电子分量是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。而电子组件是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。而电子组件是从总能量和电子密度计算得出的。为了解决使用DFT计算极化子特性的最引人注目的挑战(这是电子定位的问题),我们建议采用选定的振动特性(例如声速,剪切模量和Grüneisen参数)的计算来表示极化子跳跃能量; 所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。和Grüneisen参数,代表极化子跳跃能量;所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。和Grüneisen参数,代表极化子跳跃能量;所有这些都源自原子间键的刚度。期望这种方法比现有方法更直接,但是需要标准化。










































 京公网安备 11010802027423号
京公网安备 11010802027423号