Computational and Theoretical Chemistry ( IF 3.0 ) Pub Date : 2020-06-01 , DOI: 10.1016/j.comptc.2020.112874 Yanyan Xu , Chenze Qi , Chen Wang
|
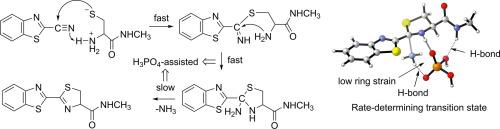
Mechanism of CBT-Cys click reaction was studied by density functional theory calculations. Calculations show that the CS bond formation followed by amino nitrogen attack pathway is the most favorable mechanism. This mechanism includes C
S bond formation, C
N bond formation, C
N bond cleavage, and several facile proton transfer steps. The rate-determining step is the C
N bond cleavage step. Except for the C
S bond formation step, all the steps occur with the assistance of phosphoric acid. The promotion of phosphoric acid on the reaction results from its strong proton-donating ability, low ring strain of the cluster structural transition states, and stabilizing effect of the hydrogen bonds in the transition states. The calculated results of the reactivities of β-mercaptoethanol, N-terminal homocysteine residues and N-terminal serine residues towards 2-cyanobenzothiazole are all consistent with the experimental observations.
中文翻译:
CBT-Cys点击反应机理的计算研究
通过密度泛函理论计算研究了CBT-Cys点击反应的机理。计算表明,C S键的形成随后是氨基氮的攻击途径是最有利的机制。该机制包括C
S键形成,C
N键形成,C
N键裂解以及几个简便的质子转移步骤。速率确定步骤是C
N键裂解步骤。除了C
在形成键的步骤中,所有步骤均在磷酸的辅助下进行。磷酸对反应的促进作用是由于其强大的质子给体能力,簇结构过渡态的低环应变以及过渡态中氢键的稳定作用。β-巯基乙醇,N末端高半胱氨酸残基和N末端丝氨酸残基对2-氰基苯并噻唑的反应性的计算结果均与实验观察结果一致。










































 京公网安备 11010802027423号
京公网安备 11010802027423号