Computational and Theoretical Chemistry ( IF 3.0 ) Pub Date : 2020-06-01 , DOI: 10.1016/j.comptc.2020.112877 Yue-Hang Dong , Zhuo Ye , Wen-Cai Lu , Yong-Xin Yao , Cai-Zhuang Wang , Kai-Ming Ho
|
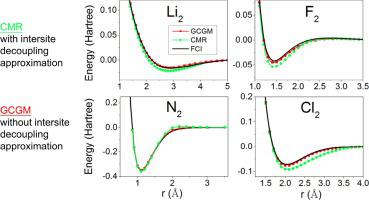
We present numerical results of ground-state energies of 9 molecules in the well-established G2 molecule set given by the Gutzwiller conjugate gradient minimization (GCGM) method. The method, beyond the commonly used Gutzwiller approximation, was recently developed based on Gutzwiller variational wave functions. We find that compared to benchmark data given by full configuration interaction, GCGM total energies are reasonably well reproduced with the minimum basis set. To include the dynamical correlation beyond the minimal basis calculations, we adopt the local density approximation for the dynamical correlation energy . By comparing the results with benchmark data given by experiments and large-basis configuration interaction, the GCGM total energies with are in general better reproduced, but discrepancies are still observed for some dimers.
中文翻译:
Gutzwiller共轭梯度最小化方法在二聚体基态能量计算中的基准
我们提供了由Gutzwiller共轭梯度最小化(GCGM)方法给出的完善的G2分子集中的9个分子的基态能量的数值结果。除了常用的Gutzwiller逼近之外,该方法最近是基于Gutzwiller变分波函数开发的。我们发现,与完全配置交互作用提供的基准数据相比,GCGM的总能量在最小基础集的情况下得到了很好的再现。为了在最小基础计算之外包括动态相关性,我们对动态相关能量采用局部密度近似。通过将结果与实验和大基数配置相互作用给出的基准数据进行比较,GCGM的总能量为 通常复制效果更好,但某些二聚体仍存在差异。










































 京公网安备 11010802027423号
京公网安备 11010802027423号