当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First-Principles Simulations of X-ray Transient Absorption for Probing Attosecond Electron Dynamics.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-05-29 , DOI: 10.1021/acs.jctc.0c00122 Min Chen 1 , Kenneth Lopata 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-05-29 , DOI: 10.1021/acs.jctc.0c00122 Min Chen 1 , Kenneth Lopata 1, 2
Affiliation
|
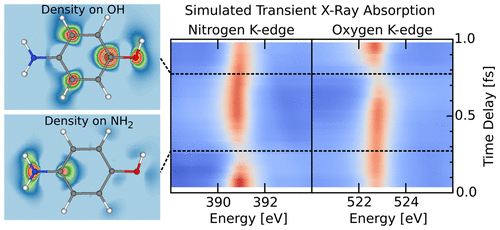
X-ray transient absorption spectroscopy (XTAS) is a promising technique for measuring electron dynamics in molecules and solids with attosecond time resolutions. In XTAS, the elemental specificity and spatial locality of core-to-valence X-ray absorption is exploited to relate modulations in the time-resolved absorption spectra to local electron density variations around particular atoms. However, interpreting these absorption modulations and frequency shifts as a function of the time delay in terms of dynamics can be challenging. In this paper, we present a first-principles study of attosecond XTAS in a selection of simple molecules based on real-time time-dependent density functional theory (RT-TDDFT) with constrained DFT to emulate the state of the system following the interaction with a ultraviolet pump laser. In general, there is a decrease in the optical density and a blue shift in the frequency with increasing electron density around the absorbing atom. In carbon monoxide (CO), modulations in the O K-edge occur at the frequency of the valence electron dynamics, while for dioxygen (O2) they occur at twice the frequency, due to the indistinguishability of the oxygen atoms. In 4-aminophenol (H2NC6H4OH), likewise, there is a decrease in the optical density and a blue shift in the frequency for the oxygen and nitrogen K-edges with increasing charge density on the O and N, respectively. Similar effects are observed in the nitrogen K-edge for a long-range charge-transfer excitation in a benzene (C6H6)–tetracyanoethylene (C6N4; TCNE) dimer but with weaker modulations due to the delocalization of the charge across the entire TCNE molecule. Additionally, in all cases, there are pre-edge features corresponding to core transitions to depopulated orbitals. These potentially offer a background-free signal that only appears in pumped molecules.
中文翻译:

用于探测阿秒电子动力学的X射线瞬态吸收的第一性原理模拟。
X射线瞬态吸收光谱(XTAS)是一种有前途的技术,可用于以亚秒级的时间分辨率测量分子和固体中的电子动力学。在XTAS中,利用了核心-价X射线吸收的元素特异性和空间局部性,将时间分辨吸收光谱中的调制与特定原子周围的局部电子密度变化相关联。然而,就动力学而言,将这些吸收调制和频移解释为时间延迟的函数可能具有挑战性。在本文中,我们提出了基于实时时变密度泛函理论(RT-TDDFT)和约束DFT来模拟简单分子选择中的阿秒XTAS的第一性原理研究,以模拟系统与相互作用后的状态。紫外线泵浦激光器。一般来说,随着吸收原子周围电子密度的增加,光密度降低,频率出现蓝移。在一氧化碳(CO)中,O K边缘的调制发生在价电子动力学频率处,而对于二氧(O2)由于氧原子的不可区分性,它们以两倍的频率出现。同样,在4-氨基苯酚(H 2 NC 6 H 4 OH)中,随着O和N电荷密度的增加,氧和氮K边缘的光密度降低,频率出现蓝移。 。对于苯(C 6 H 6)-四氰基乙烯(C 6 N 4)中的长距离电荷转移激发,在氮K边缘观察到了相似的作用; TCNE)二聚体,但由于整个TCNE分子上电荷的离域作用,其调制较弱。另外,在所有情况下,都有对应于核心过渡到人口减少的轨道的边缘特征。这些潜在地提供了仅在泵浦分子中出现的无背景信号。
更新日期:2020-07-14
中文翻译:
用于探测阿秒电子动力学的X射线瞬态吸收的第一性原理模拟。
X射线瞬态吸收光谱(XTAS)是一种有前途的技术,可用于以亚秒级的时间分辨率测量分子和固体中的电子动力学。在XTAS中,利用了核心-价X射线吸收的元素特异性和空间局部性,将时间分辨吸收光谱中的调制与特定原子周围的局部电子密度变化相关联。然而,就动力学而言,将这些吸收调制和频移解释为时间延迟的函数可能具有挑战性。在本文中,我们提出了基于实时时变密度泛函理论(RT-TDDFT)和约束DFT来模拟简单分子选择中的阿秒XTAS的第一性原理研究,以模拟系统与相互作用后的状态。紫外线泵浦激光器。一般来说,随着吸收原子周围电子密度的增加,光密度降低,频率出现蓝移。在一氧化碳(CO)中,O K边缘的调制发生在价电子动力学频率处,而对于二氧(O2)由于氧原子的不可区分性,它们以两倍的频率出现。同样,在4-氨基苯酚(H 2 NC 6 H 4 OH)中,随着O和N电荷密度的增加,氧和氮K边缘的光密度降低,频率出现蓝移。 。对于苯(C 6 H 6)-四氰基乙烯(C 6 N 4)中的长距离电荷转移激发,在氮K边缘观察到了相似的作用; TCNE)二聚体,但由于整个TCNE分子上电荷的离域作用,其调制较弱。另外,在所有情况下,都有对应于核心过渡到人口减少的轨道的边缘特征。这些潜在地提供了仅在泵浦分子中出现的无背景信号。










































 京公网安备 11010802027423号
京公网安备 11010802027423号