当前位置:
X-MOL 学术
›
Clin. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
MECP2 mutation spectrum and its clinical characteristics in a Chinese cohort.
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-05-30 , DOI: 10.1111/cge.13790 Yongxin Wen 1 , Jiaping Wang 1 , Qingping Zhang 1 , Yan Chen 1 , Xiru Wu 1 , Xinhua Bao 1
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-05-30 , DOI: 10.1111/cge.13790 Yongxin Wen 1 , Jiaping Wang 1 , Qingping Zhang 1 , Yan Chen 1 , Xiru Wu 1 , Xinhua Bao 1
Affiliation
|
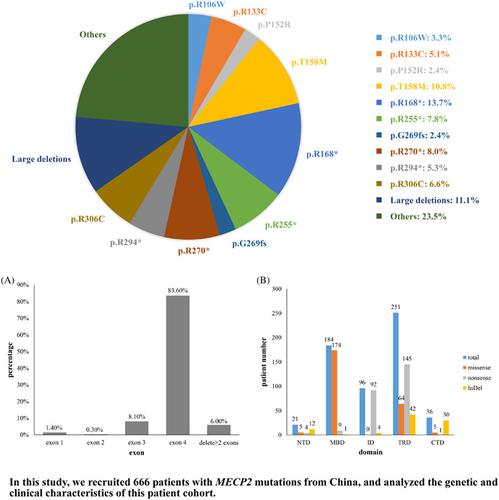
The dysfunction of methyl‐CpG‐binding protein 2 (MeCP2) is associated with several neurological disorders, of which Rett syndrome (RTT) is the most prominent. This study focused on a Chinese patient cohort with MECP2 mutations, and analyzed the characteristics of these mutations and their clinical manifestations. In total, 666 patients were identified with 126 different MECP2 mutations, including 22 novel mutations. Over 80% of patients carried an MECP2 mutation on exon 4. Nonsense and missense mutations were the most commonly reported types. Missense mutations were mainly located on methyl‐CpG‐binding domain (MBD), and nonsense mutations predominantly occurred on transcription repression domain (TRD) and inter domain. The predilection site of large deletion was exon 3 and/or exon 4. Patients with p.R133C, p.R294*, p.R306C, and C‐terminal domain (CTD) deletions were less severely affected. Significant differences were found in ambulation ability, hand function, and language among different mutation groups. Three female patients with MECP2 mutations (1 with p.R306P and 2 with p.R309W) only presented with intellectual disability/developmental delay (ID/DD), and no obvious RTT symptoms were reported. Eight male individuals with MECP2 mutations were also identified in this study, including 2 diagnosed with typical RTT, 3 with atypical RTT and 3 with ID/DD.
更新日期:2020-05-30










































 京公网安备 11010802027423号
京公网安备 11010802027423号