当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Multistate QM/QM Extrapolation of UV/Vis Absorption Spectra with Point Charge Embedding.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-05-27 , DOI: 10.1021/acs.jctc.0c00339 Kaihua Zhang 1 , Sijin Ren 1 , Marco Caricato 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-05-27 , DOI: 10.1021/acs.jctc.0c00339 Kaihua Zhang 1 , Sijin Ren 1 , Marco Caricato 1
Affiliation
|
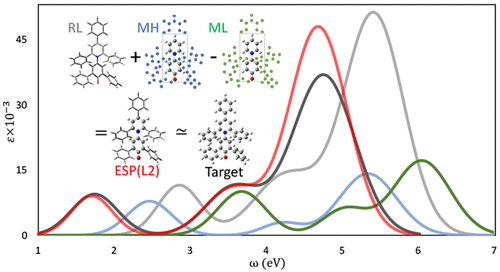
The simulation of UV/vis absorption spectra of large chromophores is prohibitively expensive with accurate quantum mechanical (QM) methods. Thus, hybrid methods, which treat the core chromophoric region at a high level of theory while the substituent effects are treated with a more computationally efficient method, may provide the best compromise between cost and accuracy. The ONIOM (Our own N-layered Integrated molecular Orbital molecular Mechanics) method has proved successful at describing ground-state processes. However, for excited states, it suffers from difficulties in matching the correct excited states among the different levels of theory. We devised an approach, based on the ONIOM extrapolation formula, to combine two QM levels of theory to extrapolate entire excitation bands rather than individual states. In this contribution, we extend the same QM/QM hybrid scheme to include polarization effects on the core region through point charge embedding. The charges are computed to reproduce the electrostatic potential of the entire chromophore at the low level of theory, with proper constraints to avoid overpolarization issues at the boundary between layers. We test this approach on a variety of model compounds that show how the multistate QM/QM-embedding scheme is able to accurately reproduce the spectrum of the entire system at the high level of theory better than (i) the bare QM/QM hybrid scheme, (ii) the low-level calculation on the entire system, and (iii) the high-level calculation on the core region.
中文翻译:

带有点电荷嵌入的UV / Vis吸收光谱的多态QM / QM外推。
使用精确的量子力学(QM)方法对大型生色团的UV / Vis吸收光谱进行模拟的成本过高。因此,以较高的理论水平处理核心发色区而用更计算有效的方法处理取代基效应的混合方法,可以在成本和准确性之间取得最佳平衡。实践证明,ONIOM(我们自己的N层集成分子轨道分子力学)方法成功地描述了基态过程。然而,对于激发态,在不同理论水平之间匹配正确的激发态存在困难。我们设计了一种基于ONIOM外推公式的方法,该方法将两个QM理论水平相结合以外推整个激发带而不是单个状态。在这项贡献中,我们扩展了相同的QM / QM混合方案,以通过点电荷嵌入对核心区域产生极化效应。计算电荷以在较低的理论水平下重现整个生色团的静电势,并带有适当的约束以避免层间边界处的过极化问题。我们在各种模型化合物上测试了这种方法,这些模型化合物显示了多态QM / QM嵌入方案如何能够以较高的理论水平准确地重现整个系统的频谱,而不是(i)裸QM / QM混合方案,(ii)整个系统的底层计算,以及(iii)核心区域的高层计算。计算电荷以在较低的理论水平下重现整个生色团的静电势,并带有适当的约束以避免层间边界处的过极化问题。我们在各种模型化合物上测试了这种方法,这些模型化合物显示了多态QM / QM嵌入方案如何能够以较高的理论水平准确地重现整个系统的频谱,而不是(i)裸QM / QM混合方案,(ii)整个系统的底层计算,以及(iii)核心区域的高层计算。计算电荷以在较低的理论水平下重现整个生色团的静电势,并带有适当的约束以避免层间边界处的过极化问题。我们在各种模型化合物上测试了这种方法,这些模型化合物显示了多态QM / QM嵌入方案如何能够以较高的理论水平准确地重现整个系统的频谱,而不是(i)裸QM / QM混合方案,(ii)整个系统的底层计算,以及(iii)核心区域的高层计算。
更新日期:2020-07-14
中文翻译:
带有点电荷嵌入的UV / Vis吸收光谱的多态QM / QM外推。
使用精确的量子力学(QM)方法对大型生色团的UV / Vis吸收光谱进行模拟的成本过高。因此,以较高的理论水平处理核心发色区而用更计算有效的方法处理取代基效应的混合方法,可以在成本和准确性之间取得最佳平衡。实践证明,ONIOM(我们自己的N层集成分子轨道分子力学)方法成功地描述了基态过程。然而,对于激发态,在不同理论水平之间匹配正确的激发态存在困难。我们设计了一种基于ONIOM外推公式的方法,该方法将两个QM理论水平相结合以外推整个激发带而不是单个状态。在这项贡献中,我们扩展了相同的QM / QM混合方案,以通过点电荷嵌入对核心区域产生极化效应。计算电荷以在较低的理论水平下重现整个生色团的静电势,并带有适当的约束以避免层间边界处的过极化问题。我们在各种模型化合物上测试了这种方法,这些模型化合物显示了多态QM / QM嵌入方案如何能够以较高的理论水平准确地重现整个系统的频谱,而不是(i)裸QM / QM混合方案,(ii)整个系统的底层计算,以及(iii)核心区域的高层计算。计算电荷以在较低的理论水平下重现整个生色团的静电势,并带有适当的约束以避免层间边界处的过极化问题。我们在各种模型化合物上测试了这种方法,这些模型化合物显示了多态QM / QM嵌入方案如何能够以较高的理论水平准确地重现整个系统的频谱,而不是(i)裸QM / QM混合方案,(ii)整个系统的底层计算,以及(iii)核心区域的高层计算。计算电荷以在较低的理论水平下重现整个生色团的静电势,并带有适当的约束以避免层间边界处的过极化问题。我们在各种模型化合物上测试了这种方法,这些模型化合物显示了多态QM / QM嵌入方案如何能够以较高的理论水平准确地重现整个系统的频谱,而不是(i)裸QM / QM混合方案,(ii)整个系统的底层计算,以及(iii)核心区域的高层计算。










































 京公网安备 11010802027423号
京公网安备 11010802027423号