当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Multidimensional Global Optimization and Robustness Analysis in the Context of Protein-Ligand Binding.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-05-25 , DOI: 10.1021/acs.jctc.0c00142 Negin Forouzesh 1 , Abhishek Mukhopadhyay 2 , Layne T Watson 1, 3, 4, 5 , Alexey V Onufriev 1, 2, 5
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-05-25 , DOI: 10.1021/acs.jctc.0c00142 Negin Forouzesh 1 , Abhishek Mukhopadhyay 2 , Layne T Watson 1, 3, 4, 5 , Alexey V Onufriev 1, 2, 5
Affiliation
|
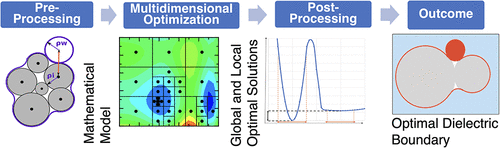
Accuracy of protein–ligand binding free energy calculations utilizing implicit solvent models is critically affected by parameters of the underlying dielectric boundary, specifically, the atomic and water probe radii. Here, a global multidimensional optimization pipeline is developed to find optimal atomic radii specifically for protein–ligand binding calculations in implicit solvent. The computational pipeline has these three key components: (1) a massively parallel implementation of a deterministic global optimization algorithm (VTDIRECT95), (2) an accurate yet reasonably fast generalized Born implicit solvent model (GBNSR6), and (3) a novel robustness metric that helps distinguish between nearly degenerate local minima via a postprocessing step of the optimization. A graph-based “kT-connectivity” approach to explore and visualize the multidimensional energy landscape is proposed: local minima that can be reached from the global minimum without exceeding a given energy threshold (kT) are considered to be connected. As an illustration of the capabilities of the optimization pipeline, we apply it to find a global optimum in the space of just five radii: four atomic (O, H, N, and C) radii and water probe radius. The optimized radii, ρW = 1.37 Å, ρC = 1.40 Å, ρH = 1.55 Å, ρN = 2.35 Å, and ρO = 1.28 Å, lead to a closer agreement of electrostatic binding free energies with the explicit solvent reference than two commonly used sets of radii previously optimized for small molecules. At the same time, the ability of the optimizer to find the global optimum reveals fundamental limits of the common two-dielectric implicit solvation model: the computed electrostatic binding free energies are still almost 4 kcal/mol away from the explicit solvent reference. The proposed computational approach opens the possibility to further improve the accuracy of practical computational protocols for binding free energy calculations.
中文翻译:

蛋白质-配体结合背景下的多维全局优化和稳健性分析。
利用隐式溶剂模型计算蛋白质-配体结合自由能的准确性受到底层介电边界参数(特别是原子和水探针半径)的严重影响。在这里,开发了一个全局多维优化管道来寻找最佳原子半径,专门用于隐式溶剂中的蛋白质-配体结合计算。计算管道具有以下三个关键组件:(1) 确定性全局优化算法 (VTDIRECT95) 的大规模并行实现,(2) 准确但相当快速的广义 Born 隐式溶剂模型 (GBNSR6),以及 (3) 新颖的鲁棒性通过优化的后处理步骤帮助区分近乎退化的局部最小值的度量。提出了一种基于图的“ kT-连通性”方法来探索和可视化多维能量景观:在不超过给定能量阈值( kT )的情况下从全局最小值达到的局部最小值被认为是连通的。作为优化管道功能的说明,我们应用它来在仅有五个半径的空间中找到全局最优值:四个原子(O、H、N 和 C)半径和水探针半径。优化后的半径 ρ W = 1.37 Å、ρ C = 1.40 Å、ρ H = 1.55 Å、ρ N = 2.35 Å 和 ρ O = 1.28 Å 使静电结合自由能与显式溶剂参考更加一致比之前针对小分子优化的两组常用半径要大。同时,优化器找到全局最优值的能力揭示了常见的双电介质隐式溶剂化模型的基本限制:计算出的静电结合自由能仍然与显式溶剂参考相差近 4 kcal/mol。所提出的计算方法为进一步提高结合自由能计算的实际计算协议的准确性提供了可能性。
更新日期:2020-07-14
中文翻译:
蛋白质-配体结合背景下的多维全局优化和稳健性分析。
利用隐式溶剂模型计算蛋白质-配体结合自由能的准确性受到底层介电边界参数(特别是原子和水探针半径)的严重影响。在这里,开发了一个全局多维优化管道来寻找最佳原子半径,专门用于隐式溶剂中的蛋白质-配体结合计算。计算管道具有以下三个关键组件:(1) 确定性全局优化算法 (VTDIRECT95) 的大规模并行实现,(2) 准确但相当快速的广义 Born 隐式溶剂模型 (GBNSR6),以及 (3) 新颖的鲁棒性通过优化的后处理步骤帮助区分近乎退化的局部最小值的度量。提出了一种基于图的“ kT-连通性”方法来探索和可视化多维能量景观:在不超过给定能量阈值( kT )的情况下从全局最小值达到的局部最小值被认为是连通的。作为优化管道功能的说明,我们应用它来在仅有五个半径的空间中找到全局最优值:四个原子(O、H、N 和 C)半径和水探针半径。优化后的半径 ρ W = 1.37 Å、ρ C = 1.40 Å、ρ H = 1.55 Å、ρ N = 2.35 Å 和 ρ O = 1.28 Å 使静电结合自由能与显式溶剂参考更加一致比之前针对小分子优化的两组常用半径要大。同时,优化器找到全局最优值的能力揭示了常见的双电介质隐式溶剂化模型的基本限制:计算出的静电结合自由能仍然与显式溶剂参考相差近 4 kcal/mol。所提出的计算方法为进一步提高结合自由能计算的实际计算协议的准确性提供了可能性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号