Bioorganic & Medicinal Chemistry ( IF 3.3 ) Pub Date : 2020-05-24 , DOI: 10.1016/j.bmc.2020.115561 Andrew P Montgomery 1 , Christopher Dobie 1 , Rémi Szabo 1 , Laura Hallam 2 , Marie Ranson 2 , Haibo Yu 2 , Danielle Skropeta 2
|
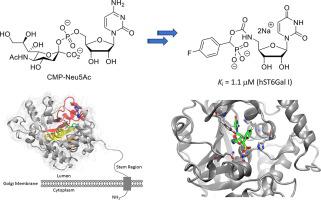
Sialic acid at the terminus of cell surface glycoconjugates is a critical element in cell-cell recognition, receptor binding and immune responses. Sialyltransferases (ST), the enzymes responsible for the biosynthesis of sialylated glycans are highly upregulated in cancer and the resulting hypersialylation of the tumour cell surface correlates strongly with tumour growth, metastasis and drug resistance. Inhibitors of human STs, in particular human ST6Gal I, are thus expected to be valuable chemical tools for the discovery of novel anticancer drugs. Herein, we report on the computationally-guided design and development of uridine-based inhibitors that replace the charged phosphodiester linker of known ST inhibitors with a neutral carbamate to improve pharmacokinetic properties and synthetic accessibility. A series of 24 carbamate-linked uridyl-based compounds were synthesised by coupling aryl and hetaryl α-hydroxyphosphonates with a 5′-amino-5′-deoxyuridine fragment. The inhibitory activities of the newly synthesised compounds against recombinant human ST6Gal I were determined using a luminescent microplate assay, and five promising inhibitors with Ki’s ranging from 1 to 20 µM were identified. These results show that carbamate-linked uridyl-based compounds are a potential new class of readily accessible, non-cytotoxic ST inhibitors to be further explored.
中文翻译:
设计,合成和评估人ST6Gal I的氨基甲酸酯连接的基于尿嘧啶的抑制剂。
在细胞表面糖缀合物末端的唾液酸是细胞-细胞识别,受体结合和免疫应答中的关键元素。唾液酸化转移酶(ST)是负责唾液酸化聚糖生物合成的酶,在癌症中高度上调,并且所产生的肿瘤细胞表面过度唾液酸化与肿瘤的生长,转移和耐药性密切相关。因此,预期人类ST,特别是人类ST6Gal I的抑制剂是发现新型抗癌药物的有价值的化学工具。本文中,我们报道了基于尿苷的抑制剂的计算指导设计和开发,该抑制剂以中性氨基甲酸酯替代已知ST抑制剂的带电磷酸二酯键,以改善药代动力学性质和合成可及性。通过将芳基和杂芳基α-羟基膦酸酯与5'-氨基-5'-脱氧尿苷片段偶联,合成了一系列24个氨基甲酸酯连接的基于尿嘧啶的化合物。使用荧光酶标仪测定了新合成的化合物对重组人ST6Gal I的抑制活性,以及五种有前途的抑制剂确定的K i范围为1至20 µM。这些结果表明,氨基甲酸酯连接的基于尿嘧啶基的化合物是潜在的一类易于获得的,无细胞毒性的ST抑制剂,有待进一步研究。










































 京公网安备 11010802027423号
京公网安备 11010802027423号