当前位置:
X-MOL 学术
›
J. Mol. Cell. Cardiol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Nuclear-mitochondrial communication involving miR-181c plays an important role in cardiac dysfunction during obesity.
Journal of Molecular and Cellular Cardiology ( IF 5 ) Pub Date : 2020-05-19 , DOI: 10.1016/j.yjmcc.2020.05.009 Barbara Roman 1 , Pawandeep Kaur 2 , Deepthi Ashok 3 , Mark Kohr 4 , Roopa Biswas 5 , Brian O'Rourke 3 , Charles Steenbergen 1 , Samarjit Das 6
Journal of Molecular and Cellular Cardiology ( IF 5 ) Pub Date : 2020-05-19 , DOI: 10.1016/j.yjmcc.2020.05.009 Barbara Roman 1 , Pawandeep Kaur 2 , Deepthi Ashok 3 , Mark Kohr 4 , Roopa Biswas 5 , Brian O'Rourke 3 , Charles Steenbergen 1 , Samarjit Das 6
Affiliation
|
AIMS
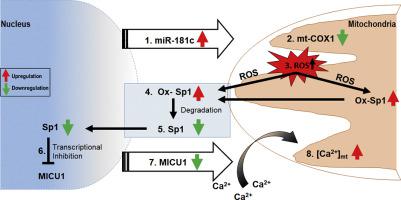
In cardiomyocytes, there is microRNA (miR) in the mitochondria that originates from the nuclear genome and matures in the cytoplasm before translocating into the mitochondria. Overexpression of one such miR, miR-181c, can lead to heart failure by stimulating reactive oxygen species (ROS) production and increasing mitochondrial calcium level ([Ca2+]m). Mitochondrial calcium uptake 1 protein (MICU1), a regulatory protein in the mitochondrial calcium uniporter complex, plays an important role in regulating [Ca2+]m. Obesity results in miR-181c overexpression and a decrease in MICU1. We hypothesize that lowering miR-181c would protect against obesity-induced cardiac dysfunction.
METHODS AND RESULTS
We used an in vivo mouse model of high-fat diet (HFD) for 18 weeks and induced high lipid load in H9c2 cells with oleate-conjugated bovine serum albumin in vitro. We tested the cardioprotective role of lowering miR-181c by using miR-181c/d-/- mice (in vivo) and AntagomiR against miR-181c (in vitro). HFD significantly upregulated heart levels of miR-181c and led to cardiac hypertrophy in wild-type mice, but not in miR-181c/d-/- mice. HFD also increased in ROS production and pyruvate dehydrogenase activity (a surrogate for [Ca2+]m), but the increases were alleviated in miR-181c/d-/- mice. Moreover, miR-181c/d-/- mice fed a HFD had higher levels of MICU1 than did wild-type mice fed a HFD, attenuating the rise in [Ca2+]m. Overexpression of miR-181c in neonatal ventricular cardiomyocytes (NMVM) caused increased ROS production, which oxidized transcription factor Sp1 and led to a loss of Sp1, thereby slowing MICU1 transcription. Hence, miR-181c increases [Ca2+]m through Sp1 oxidation and downregulation of MICU1, suggesting that the cardioprotective effect of miR-181c/d-/- results from inhibition of Sp1 oxidation.
CONCLUSION
This study has identified a unique nuclear-mitochondrial communication mechanism in the heart orchestrated by miR-181c. Obesity-induced overexpression of miR-181c increases [Ca2+]m via downregulation of MICU1 and leads to cardiac injury. A strategy to inhibit miR-181c in cardiomyocytes can preserve cardiac function during obesity by improving mitochondrial function. Altering miR-181c expression may provide a pharmacologic approach to improve cardiomyopathy in individuals with obesity/type 2 diabetes.
中文翻译:

涉及 miR-181c 的核-线粒体通讯在肥胖期间的心脏功能障碍中起重要作用。
目的在心肌细胞中,线粒体中有微小RNA(miR),其起源于核基因组,并在细胞质中成熟,然后转移到线粒体中。其中一种 miR,miR-181c 的过表达可通过刺激活性氧 (ROS) 产生和增加线粒体钙水平 ([Ca2+]m) 导致心力衰竭。线粒体钙摄取 1 蛋白 (MICU1) 是线粒体钙单向转运体复合物中的一种调节蛋白,在调节 [Ca2+]m 中起重要作用。肥胖导致 miR-181c 过表达和 MICU1 减少。我们假设降低 miR-181c 可以防止肥胖引起的心功能不全。方法和结果 我们使用高脂饮食 (HFD) 的体内小鼠模型 18 周,并在体外用油酸结合牛血清白蛋白诱导 H9c2 细胞的高脂质负荷。我们通过使用 miR-181c/d-/- 小鼠(体内)和针对 miR-181c(体外)的 AntagomiR 测试了降低 miR-181c 的心脏保护作用。HFD 显着上调 miR-181c 的心脏水平并导致野生型小鼠的心脏肥大,但在 miR-181c/d-/- 小鼠中没有。HFD 还增加了 ROS 的产生和丙酮酸脱氢酶活性([Ca2+]m 的替代物),但在 miR-181c/d-/- 小鼠中这种增加有所缓解。此外,喂食 HFD 的 miR-181c/d-/- 小鼠的 MICU1 水平高于喂食 HFD 的野生型小鼠,从而减弱了 [Ca2+]m 的升高。miR-181c 在新生儿心室心肌细胞 (NMVM) 中的过表达导致 ROS 产生增加,其氧化转录因子Sp1并导致Sp1丢失,从而减慢MICU1转录。因此,miR-181c 通过 Sp1 氧化和 MICU1 的下调增加 [Ca2+]m,表明 miR-181c/d-/- 的心脏保护作用是由抑制 Sp1 氧化引起的。结论 本研究确定了由 miR-181c 协调的心脏中独特的核-线粒体通讯机制。肥胖诱导的 miR-181c 过表达通过下调 MICU1 增加 [Ca2+]m 并导致心脏损伤。抑制心肌细胞中 miR-181c 的策略可以通过改善线粒体功能来保护肥胖期间的心脏功能。改变 miR-181c 表达可能提供一种药理学方法来改善肥胖/2 型糖尿病患者的心肌病。
更新日期:2020-05-19
中文翻译:
涉及 miR-181c 的核-线粒体通讯在肥胖期间的心脏功能障碍中起重要作用。
目的在心肌细胞中,线粒体中有微小RNA(miR),其起源于核基因组,并在细胞质中成熟,然后转移到线粒体中。其中一种 miR,miR-181c 的过表达可通过刺激活性氧 (ROS) 产生和增加线粒体钙水平 ([Ca2+]m) 导致心力衰竭。线粒体钙摄取 1 蛋白 (MICU1) 是线粒体钙单向转运体复合物中的一种调节蛋白,在调节 [Ca2+]m 中起重要作用。肥胖导致 miR-181c 过表达和 MICU1 减少。我们假设降低 miR-181c 可以防止肥胖引起的心功能不全。方法和结果 我们使用高脂饮食 (HFD) 的体内小鼠模型 18 周,并在体外用油酸结合牛血清白蛋白诱导 H9c2 细胞的高脂质负荷。我们通过使用 miR-181c/d-/- 小鼠(体内)和针对 miR-181c(体外)的 AntagomiR 测试了降低 miR-181c 的心脏保护作用。HFD 显着上调 miR-181c 的心脏水平并导致野生型小鼠的心脏肥大,但在 miR-181c/d-/- 小鼠中没有。HFD 还增加了 ROS 的产生和丙酮酸脱氢酶活性([Ca2+]m 的替代物),但在 miR-181c/d-/- 小鼠中这种增加有所缓解。此外,喂食 HFD 的 miR-181c/d-/- 小鼠的 MICU1 水平高于喂食 HFD 的野生型小鼠,从而减弱了 [Ca2+]m 的升高。miR-181c 在新生儿心室心肌细胞 (NMVM) 中的过表达导致 ROS 产生增加,其氧化转录因子Sp1并导致Sp1丢失,从而减慢MICU1转录。因此,miR-181c 通过 Sp1 氧化和 MICU1 的下调增加 [Ca2+]m,表明 miR-181c/d-/- 的心脏保护作用是由抑制 Sp1 氧化引起的。结论 本研究确定了由 miR-181c 协调的心脏中独特的核-线粒体通讯机制。肥胖诱导的 miR-181c 过表达通过下调 MICU1 增加 [Ca2+]m 并导致心脏损伤。抑制心肌细胞中 miR-181c 的策略可以通过改善线粒体功能来保护肥胖期间的心脏功能。改变 miR-181c 表达可能提供一种药理学方法来改善肥胖/2 型糖尿病患者的心肌病。


























 京公网安备 11010802027423号
京公网安备 11010802027423号