当前位置:
X-MOL 学术
›
Clin. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Refinement of the clinical and mutational spectrum of UBE2A deficiency syndrome.
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-05-15 , DOI: 10.1111/cge.13775 Viviana Cordeddu 1 , Erica L Macke 2 , Francesca Clementina Radio 3 , Stefania Lo Cicero 4 , Francesca Pantaleoni 3 , Massimo Tatti 4 , Emanuele Bellacchio 3 , Andrea Ciolfi 3 , Emanuele Agolini 3 , Alessandro Bruselles 4 , Nicola Brunetti-Pierri 5, 6 , Mohnish Suri 7 , Katherine S Josephs 8 , Meriel McEntagart 8 , Brendan Lanpher 9 , Katherine C Nickels 10 , Andrea Haworth 11 , Laura Reed 11 , Gerarda Cappuccio 5, 6 , Isabella Mammi 12 , Jessica M Tarnowski 9 , Antonio Novelli 3 , 13 , Daniela Melis 14 , Bert Callewaert 15, 16 , Bruno Dallapiccola 3 , Eric Klee 2, 9 , Marco Tartaglia 3
Clinical Genetics ( IF 2.9 ) Pub Date : 2020-05-15 , DOI: 10.1111/cge.13775 Viviana Cordeddu 1 , Erica L Macke 2 , Francesca Clementina Radio 3 , Stefania Lo Cicero 4 , Francesca Pantaleoni 3 , Massimo Tatti 4 , Emanuele Bellacchio 3 , Andrea Ciolfi 3 , Emanuele Agolini 3 , Alessandro Bruselles 4 , Nicola Brunetti-Pierri 5, 6 , Mohnish Suri 7 , Katherine S Josephs 8 , Meriel McEntagart 8 , Brendan Lanpher 9 , Katherine C Nickels 10 , Andrea Haworth 11 , Laura Reed 11 , Gerarda Cappuccio 5, 6 , Isabella Mammi 12 , Jessica M Tarnowski 9 , Antonio Novelli 3 , 13 , Daniela Melis 14 , Bert Callewaert 15, 16 , Bruno Dallapiccola 3 , Eric Klee 2, 9 , Marco Tartaglia 3
Affiliation
|
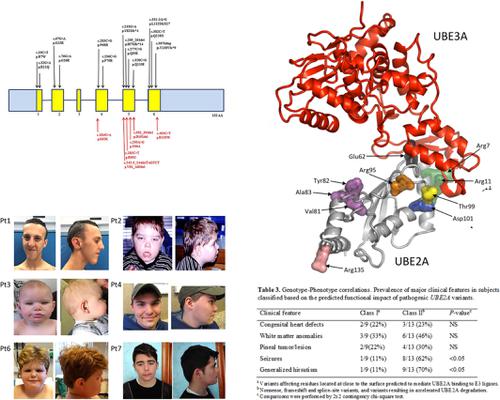
UBE2A deficiency, that is, intellectual disability (ID) Nascimento type (MIM 300860), is an X‐linked syndrome characterized by developmental delay, moderate to severe ID, seizures, dysmorphisms, skin anomalies, and urogenital malformations. Forty affected subjects have been reported thus far, with 31 cases having intragenic UBE2A variants. Here, we report on additional eight affected subjects from seven unrelated families who were found to be hemizygous for previously unreported UBE2A missense variants (p.Glu62Lys, p.Arg95Cys, p.Thr99Ala, and p.Arg135Trp) or small in‐frame deletions (p.Val81_Ala83del, and p.Asp101del). A wide phenotypic spectrum was documented in these subjects, ranging from moderate ID associated with mild dysmorphisms to severe features including congenital heart defects (CHD), severe cognitive impairment, and pineal gland tumors. Four variants affected residues (Glu62, Arg95, Thr99 and Asp101) that contribute to stabilizing the structure of the E3 binding domain. The three‐residue in‐frame deletion, p.Val81_Ala83del, resulted from aberrant processing of the transcript. This variant and p.Arg135Trp mapped to regions of the protein located far from the E3 binding region, and caused variably accelerated protein degradation. By reviewing available clinical information, we revise the clinical and molecular profile of the disorder and document genotype‐phenotype correlations. Pineal gland cysts/tumors, CHD and hypogammaglobulinemia emerge as recurrent features.
中文翻译:

完善UBE2A缺乏症候群的临床和突变谱。
UBE2A缺乏症,即智力障碍(ID)纳西门托型(MIM 300860),是一种X连锁综合征,其特征在于发育延迟,中度至重度ID,癫痫发作,畸形,皮肤异常和泌尿生殖器畸形。迄今为止,已报道了40名受累受试者,其中31例具有基因内UBE2A变异。在这里,我们报告了来自七个无关家庭的另外八名受影响的受试者,这些受试者被发现以前未报告的UBE2A是半合子的错义变体(p.Glu62Lys,p.Arg95Cys,p.Thr99Ala和p.Arg135Trp)或较小的框内缺失(p.Val81_Ala83del和p.Asp101del)。这些受试者记录了广泛的表型谱,范围从与轻度畸形相关的中度ID到包括先天性心脏缺陷(CHD),严重的认知障碍和松果体肿瘤在内的严重特征。四个变体影响残基(Glu62,Arg95,Thr99和Asp101),这些残基有助于稳定E3结合域的结构。三残基框内缺失,p.Val81_Ala83del,是由于转录本的异常处理所致。该变体和p.Arg135Trp定位于远离E3结合区的蛋白质区域,并导致蛋白质降解加速变化。通过查看可用的临床信息,我们修订了该疾病的临床和分子特征,并记录了基因型与表型的相关性。松果体囊肿/肿瘤,冠心病和低球蛋白血症是复发的特征。
更新日期:2020-07-15
中文翻译:
完善UBE2A缺乏症候群的临床和突变谱。
UBE2A缺乏症,即智力障碍(ID)纳西门托型(MIM 300860),是一种X连锁综合征,其特征在于发育延迟,中度至重度ID,癫痫发作,畸形,皮肤异常和泌尿生殖器畸形。迄今为止,已报道了40名受累受试者,其中31例具有基因内UBE2A变异。在这里,我们报告了来自七个无关家庭的另外八名受影响的受试者,这些受试者被发现以前未报告的UBE2A是半合子的错义变体(p.Glu62Lys,p.Arg95Cys,p.Thr99Ala和p.Arg135Trp)或较小的框内缺失(p.Val81_Ala83del和p.Asp101del)。这些受试者记录了广泛的表型谱,范围从与轻度畸形相关的中度ID到包括先天性心脏缺陷(CHD),严重的认知障碍和松果体肿瘤在内的严重特征。四个变体影响残基(Glu62,Arg95,Thr99和Asp101),这些残基有助于稳定E3结合域的结构。三残基框内缺失,p.Val81_Ala83del,是由于转录本的异常处理所致。该变体和p.Arg135Trp定位于远离E3结合区的蛋白质区域,并导致蛋白质降解加速变化。通过查看可用的临床信息,我们修订了该疾病的临床和分子特征,并记录了基因型与表型的相关性。松果体囊肿/肿瘤,冠心病和低球蛋白血症是复发的特征。










































 京公网安备 11010802027423号
京公网安备 11010802027423号