Bioorganic & Medicinal Chemistry ( IF 3.3 ) Pub Date : 2020-05-06 , DOI: 10.1016/j.bmc.2020.115544 Lalit K Golani 1 , Farhana Islam 1 , Carrie O'Connor 2 , Aamod S Dekhne 2 , Zhanjun Hou 3 , Larry H Matherly 4 , Aleem Gangjee 1
|
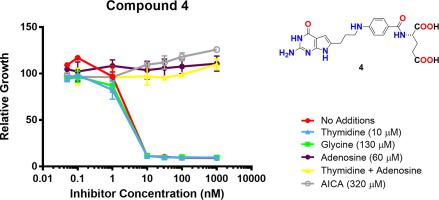
Tumor-targeted 6-substituted pyrrolo[2,3-d]pyrimidine benzoyl compounds based on 2 were isosterically modified at the 4-carbon bridge by replacing the vicinal (C11) carbon by heteroatoms N (4), O (5) or S (6), or with an N-substituted formyl (7), trifluoroacetyl (8) or acetyl (9). Replacement with sulfur (6) afforded the most potent KB tumor cell inhibitor, ~6-fold better than the parent 2. In addition, 6 retained tumor transport selectivity via folate receptor (FR) α and -β over the ubiquitous reduced folate carrier (RFC). FRα-mediated cell inhibition for 6 was generally equivalent to 2, while the FRβ-mediated activity was improved by 16-fold over 2. N (4) and O (5) substitutions afforded similar tumor cell inhibitions as 2, with selectivity for FRα and -β over RFC. The N-substituted analogs 7–9 also preserved transport selectivity for FRα and -β over RFC. For FRα-expressing CHO cells, potencies were in the order of 8 > 7 > 9. Whereas 8 and 9 showed similar results with FRβ-expressing CHO cells, 7 was ~16-fold more active than 2. By nucleoside rescue experiments, all the compounds inhibited de novo purine biosynthesis, likely at the step catalyzed by glycinamide ribonucleotide formyltransferase. Thus, heteroatom replacements of the CH2 in the bridge of 2 afford analogs with increased tumor cell inhibition that could provide advantages over 2, as well as tumor transport selectivity over clinically used antifolates including methotrexate and pemetrexed.
中文翻译:
设计,合成和生物学评价新型吡咯并[2,3-d]嘧啶作为肿瘤靶向剂,对还原性叶酸载体具有高亲和力的叶酸受体选择性吸收肿瘤。
在2碳桥上,通过杂原子N(4),O(5)或S取代邻位(C11)碳,在4碳桥上等位修饰基于肿瘤的6取代吡咯并[2,3- d ]嘧啶苯甲酰基化合物。(6),或具有N-取代的甲酰基(7),三氟乙酰基(8)或乙酰基(9)。用硫替代(6)提供了最有效的KB肿瘤细胞抑制剂,比亲代2好约6倍。另外6通过叶酸受体(FR)α和-β在普遍存在的还原性叶酸载体(RFC)上保留了肿瘤的转运选择性。FRα介导的6细胞抑制作用通常相当于2,而FRβ介导的活性是2的16倍。N(4)和O(5)替代提供了与2类似的肿瘤细胞抑制作用,相对RFC对FRα和-β具有选择性。所述Ñ取代类似物7 - 9为FRα还保存运输选择性和-β在RFC。对于表达FRα的CHO细胞,效价为8 > 7 > 9。而8和9在表达FRβ的CHO细胞中显示出相似的结果,而7的活性是2的16倍。通过核苷挽救实验,所有化合物均可能在嘌呤酰胺核糖核苷酸甲酰基转移酶催化的步骤中抑制嘌呤从头进行生物合成。因此,CH的杂原子替换2中的桥2得到与增加的肿瘤细胞的生长抑制,可以提供超过优点类似物2,以及肿瘤运输选择性超过临床使用的抗叶酸剂包括甲氨蝶呤和培美曲塞。










































 京公网安备 11010802027423号
京公网安备 11010802027423号