Current Topics in Medicinal Chemistry ( IF 2.9 ) Pub Date : 2020-06-30 , DOI: 10.2174/1568026620666200502005853 Prashant S Kharkar 1
|
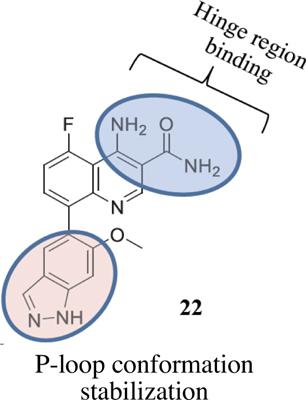
Kinases remain one of the major attractive therapeutic targets for a large number of indications such as cancer, rheumatoid arthritis, cardiac failure and many others. Design and development of kinase inhibitors (ATP-competitive, allosteric or covalent) is a clinically validated and successful strategy in the pharmaceutical industry. The perks come with limitations, particularly the development of resistance to highly potent and selective inhibitors. When this happens, the cycle needs to be repeated, i.e., the design and development of kinase inhibitors active against the mutated forms. The complexity of tumor milieu makes it awfully difficult for these molecularly-targeted therapies to work. Every year newer and better versions of these agents are introduced in the clinic. Several computational approaches such as structure-, ligand-based or hybrid ones continue to live up to their potential in discovering novel kinase inhibitors. New schools of thought in this area continue to emerge, e.g., development of dual-target kinase inhibitors. But there are fundamental issues with this approach. It is indeed difficult to selectively optimize binding at two entirely different or related kinases. In addition to the conventional strategies, modern technologies (machine learning, deep learning, artificial intelligence, etc.) started yielding the results and building success stories. Computational tools invariably played a critical role in catalysing the phenomenal progress in kinase drug discovery field. The present review summarized the progress in utilizing computational methods and tools for discovering (mutant-)selective tyrosine kinase inhibitor drugs in the last three years (2017-2019). Representative investigations have been discussed, while others are merely listed. The author believes that the enthusiastic reader will be inspired to dig out the cited literature extensively to appreciate the progress made so far and the future prospects of the field.
中文翻译:
设计(突变型)酪氨酸激酶抑制剂的计算方法:最新技术和未来前景。
对于许多适应症例如癌症,类风湿性关节炎,心力衰竭和许多其他适应症,激酶仍然是主要的有吸引力的治疗靶标之一。激酶抑制剂(ATP竞争性,变构或共价)抑制剂的设计和开发是制药业经过临床验证且成功的策略。这些特权具有局限性,特别是对高效和选择性抑制剂的抗性发展。当发生这种情况时,需要重复该循环,即,设计和开发对突变形式具有活性的激酶抑制剂。肿瘤环境的复杂性使这些分子靶向疗法难以发挥作用。每年,诊所都会引入这些代理的更新更好的版本。几种计算方法,例如结构,基于配体的或杂合的继续发挥其发现新型激酶抑制剂的潜力。该领域的新思想不断涌现,例如开发双靶激酶抑制剂。但是这种方法存在一些基本问题。确实很难选择性优化两种完全不同或相关激酶的结合。除了常规策略外,现代技术(机器学习,深度学习,人工智能等)也开始产生成果并建立成功案例。计算工具始终在催化激酶药物发现领域的惊人进展中起着至关重要的作用。本综述总结了过去三年(2017-2019年)利用计算方法和工具发现(突变)选择性酪氨酸激酶抑制剂药物的进展。讨论了代表性调查,仅列出了其他调查。作者认为,热情的读者将受到启发,以广泛地挖掘引用的文献,以欣赏迄今为止所取得的进展以及该领域的未来前景。









































 京公网安备 11010802027423号
京公网安备 11010802027423号