当前位置:
X-MOL 学术
›
Fluid Phase Equilibr.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Molecular dynamics simulation on Na+−F− ion-pair association from ambient to supercritical water
Fluid Phase Equilibria ( IF 2.6 ) Pub Date : 2020-07-01 , DOI: 10.1016/j.fluid.2020.112615 Wei Zhang , Tinggui Yan
Fluid Phase Equilibria ( IF 2.6 ) Pub Date : 2020-07-01 , DOI: 10.1016/j.fluid.2020.112615 Wei Zhang , Tinggui Yan
|
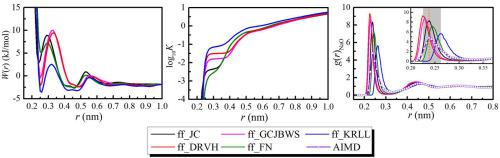
Abstract Constrained classical molecular dynamics simulations were carried out to investigate the sodium fluoride associations in dilute aqueous solutions from ambient to supercritical thermodynamic condition. Solvent mediated potential of mean force (PMF) of Na+−F− ions pair was calculated by thermodynamic integration with five different force fields and the SPC/E water model. The corresponding association constants were also calculated from the PMFs. The structure and dynamics of Na+ and F− in water calculated from these force fields were compared with the results from ab initio molecular dynamics at 298 K. The results show Na+−F− ion-pair tends to associate with increasing temperature and dissociate with increasing pressure. The association constants of the ion-pairs (Kall) including contact ion-pair (CIP), solvent shared ion-pair (SShIP) and solvent separated ion-pair (SSIP) from different force fields agreed well with each other within 0.4 log unit in the whole thermodynamic condition, and they are also in accordance with the recently published data at 298 K. But in the sub-supercritical region, significant deviations occurred between the contact ion-pair association constants (KCIP) and Kall for each force field and also between KCIP from different force fields. These two kinds of deviations decreased at higher temperature when CIP became the predominated configuration. Comparison of radial distribution functions (RDFs) characteristics from the five force fields with that from AIMD calculation shows that the force field parameters developed by combing single ion properties with mineral lattice energy or ion-pair interaction properties is better.
中文翻译:

从环境水到超临界水的 Na+−F− 离子对缔合的分子动力学模拟
摘要 进行了受约束的经典分子动力学模拟,以研究从环境到超临界热力学条件的稀水溶液中氟化钠的缔合。Na+-F- 离子对的平均力 (PMF) 的溶剂介导电位是通过热力学积分与五个不同的力场和 SPC/E 水模型计算的。相应的缔合常数也由 PMF 计算。将这些力场计算出的水中 Na+ 和 F- 的结构和动力学与 298 K 从头分子动力学的结果进行比较。 结果表明 Na+-F- 离子对倾向于与温度升高相关并随着温度升高而解离压力。离子对(Kall)的结合常数,包括接触离子对(CIP),来自不同力场的溶剂共享离子对(SShIP)和溶剂分离离子对(SSIP)在整个热力学条件下在0.4 log单位内相互吻合,并且它们也与最近公布的298 K数据一致但是在亚超临界区,每个力场的接触离子对结合常数(KCIP)和Kall之间以及来自不同力场的KCIP之间发生了显着的偏差。当 CIP 成为主要配置时,这两种偏差在较高温度下减小。五个力场的径向分布函数(RDF)特性与AIMD计算的径向分布函数(RDF)特性的比较表明,通过结合单离子特性与矿物晶格能或离子对相互作用特性得到的力场参数更好。
更新日期:2020-07-01
中文翻译:
从环境水到超临界水的 Na+−F− 离子对缔合的分子动力学模拟
摘要 进行了受约束的经典分子动力学模拟,以研究从环境到超临界热力学条件的稀水溶液中氟化钠的缔合。Na+-F- 离子对的平均力 (PMF) 的溶剂介导电位是通过热力学积分与五个不同的力场和 SPC/E 水模型计算的。相应的缔合常数也由 PMF 计算。将这些力场计算出的水中 Na+ 和 F- 的结构和动力学与 298 K 从头分子动力学的结果进行比较。 结果表明 Na+-F- 离子对倾向于与温度升高相关并随着温度升高而解离压力。离子对(Kall)的结合常数,包括接触离子对(CIP),来自不同力场的溶剂共享离子对(SShIP)和溶剂分离离子对(SSIP)在整个热力学条件下在0.4 log单位内相互吻合,并且它们也与最近公布的298 K数据一致但是在亚超临界区,每个力场的接触离子对结合常数(KCIP)和Kall之间以及来自不同力场的KCIP之间发生了显着的偏差。当 CIP 成为主要配置时,这两种偏差在较高温度下减小。五个力场的径向分布函数(RDF)特性与AIMD计算的径向分布函数(RDF)特性的比较表明,通过结合单离子特性与矿物晶格能或离子对相互作用特性得到的力场参数更好。


























 京公网安备 11010802027423号
京公网安备 11010802027423号