当前位置:
X-MOL 学术
›
Mol. Genet. Genomic Med.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Spinal muscular atrophy caused by a novel Alu-mediated deletion of exons 2a-5 in SMN1 undetectable with routine genetic testing.
Molecular Genetics & Genomic Medicine ( IF 1.5 ) Pub Date : 2020-04-26 , DOI: 10.1002/mgg3.1238 Ivana Jedličková 1 , Anna Přistoupilová 1 , Lenka Nosková 1 , Filip Majer 1 , Viktor Stránecký 1 , Hana Hartmannová 1 , Kateřina Hodaňová 1 , Helena Trešlová 1 , Michaela Hýblová 2 , Peter Solár 3 , Gabriel Minárik 2 , Mária Giertlová 2 , Stanislav Kmoch 1
Molecular Genetics & Genomic Medicine ( IF 1.5 ) Pub Date : 2020-04-26 , DOI: 10.1002/mgg3.1238 Ivana Jedličková 1 , Anna Přistoupilová 1 , Lenka Nosková 1 , Filip Majer 1 , Viktor Stránecký 1 , Hana Hartmannová 1 , Kateřina Hodaňová 1 , Helena Trešlová 1 , Michaela Hýblová 2 , Peter Solár 3 , Gabriel Minárik 2 , Mária Giertlová 2 , Stanislav Kmoch 1
Affiliation
|
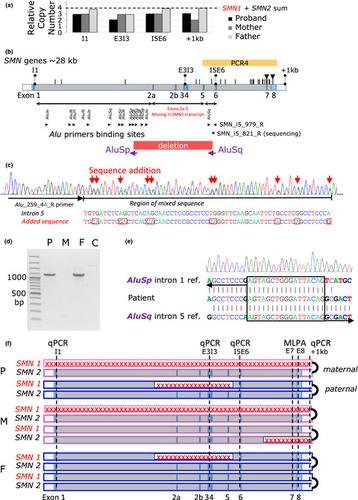
Spinal muscular atrophy (SMA) is an inherited neuromuscular disease affecting 1 in 8,000 newborns. The majority of patients carry bi‐allelic variants in the survival of motor neuron 1 gene (SMN1 ). SMN1 is located in a duplicated region on chromosome 5q13 that contains Alu elements and is predisposed to genomic rearrangements. Due to the genomic complexity of the SMN region and genetic heterogeneity, approximately 50% of SMA patients remain without genetic diagnosis that is a prerequisite for genetic treatments. In this work we describe the diagnostic odyssey of one SMA patient in whom routine diagnostics identified only a maternal heterozygous SMN1Δ(7–8) deletion.
中文翻译:

常规遗传学检测无法检测到SMN1中新的Alu介导的外显子2a-5缺失引起的脊髓性肌萎缩。
脊髓性肌萎缩症(SMA)是一种遗传性神经肌肉疾病,影响8,000名新生儿中的1名。大多数患者在运动神经元1基因(SMN1)的存活中带有双等位基因变异。SMN1位于染色体5q13上的一个重复区域中,该区域包含Alu元素,并且容易发生基因组重排。由于SMN地区的基因组复杂性和遗传异质性,大约50%的SMA患者没有进行遗传诊断,而遗传诊断是遗传治疗的先决条件。在这项工作中,我们描述了一名SMA患者的诊断经历,其中常规诊断仅发现了母体杂合SMN1Δ(7-8)缺失。
更新日期:2020-04-26
中文翻译:
常规遗传学检测无法检测到SMN1中新的Alu介导的外显子2a-5缺失引起的脊髓性肌萎缩。
脊髓性肌萎缩症(SMA)是一种遗传性神经肌肉疾病,影响8,000名新生儿中的1名。大多数患者在运动神经元1基因(SMN1)的存活中带有双等位基因变异。SMN1位于染色体5q13上的一个重复区域中,该区域包含Alu元素,并且容易发生基因组重排。由于SMN地区的基因组复杂性和遗传异质性,大约50%的SMA患者没有进行遗传诊断,而遗传诊断是遗传治疗的先决条件。在这项工作中,我们描述了一名SMA患者的诊断经历,其中常规诊断仅发现了母体杂合SMN1Δ(7-8)缺失。










































 京公网安备 11010802027423号
京公网安备 11010802027423号