Cell Systems ( IF 9.0 ) Pub Date : 2020-04-22 , DOI: 10.1016/j.cels.2020.04.001 Gryte Satas 1, 2 , Simone Zaccaria 2 , Geoffrey Mon 2 , Benjamin J Raphael 2
|
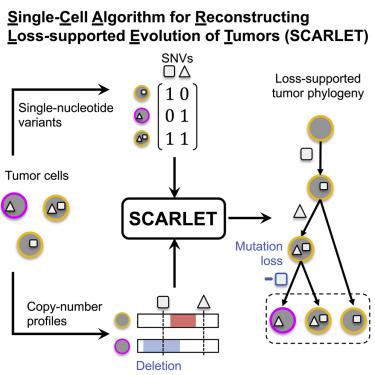
A small number of somatic mutations drive the development of cancer, but all somatic mutations are markers of the evolutionary history of a tumor. Prominent methods to construct phylogenies from single-cell-sequencing data use single-nucleotide variants (SNVs) as markers but fail to adequately account for copy-number aberrations (CNAs), which can overlap SNVs and result in SNV losses. Here, we introduce SCARLET, an algorithm that infers tumor phylogenies from single-cell DNA sequencing data while accounting for both CNA-driven loss of SNVs and sequencing errors. SCARLET outperforms existing methods on simulated data, with more accurate inference of the order in which mutations were acquired and the mutations present in individual cells. Using a single-cell dataset from a patient with colorectal cancer, SCARLET constructs a tumor phylogeny that is consistent with the observed CNAs and suggests an alternate origin for the patient’s metastases. SCARLET is available at: github.com/raphael-group/scarlet.
中文翻译:
SCARLET:具有拷贝数限制突变丢失的单细胞肿瘤系统发育推断。
少数体细胞突变驱动癌症的发展,但所有体细胞突变都是肿瘤进化史的标志。从单细胞测序数据构建系统发育的重要方法使用单核苷酸变异(SNV)作为标记,但无法充分考虑拷贝数畸变(CNA),拷贝数畸变可能与 SNV 重叠并导致 SNV 丢失。在这里,我们介绍 SCARLET,这是一种从单细胞 DNA 测序数据推断肿瘤系统发育的算法,同时考虑到 CNA 驱动的 SNV 丢失和测序错误。 SCARLET 在模拟数据方面优于现有方法,可以更准确地推断获得突变的顺序以及单个细胞中存在的突变。 SCARLET 使用结直肠癌患者的单细胞数据集构建了与观察到的 CNAs 一致的肿瘤系统发育,并提出了患者转移的替代起源。 SCARLET 可在以下位置获取:github.com/raphael-group/scarlet。










































 京公网安备 11010802027423号
京公网安备 11010802027423号