当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Catalytic Asymmetric Reactions with N-Metallated Azomethine Ylides.
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2020-04-22 , DOI: 10.1021/acs.accounts.0c00113 Liang Wei 1 , Xin Chang 1 , Chun-Jiang Wang 1, 2
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2020-04-22 , DOI: 10.1021/acs.accounts.0c00113 Liang Wei 1 , Xin Chang 1 , Chun-Jiang Wang 1, 2
Affiliation
|
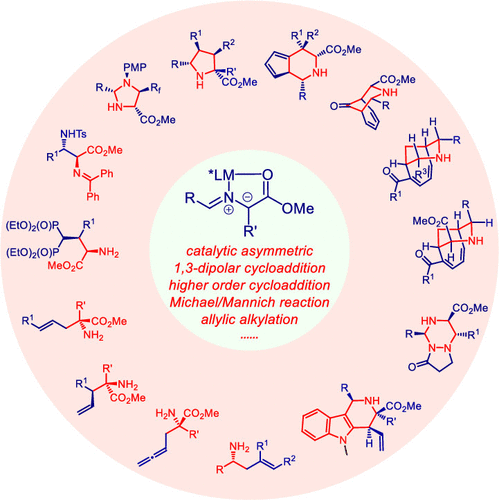
ConspectusOptically active nitrogen-containing compounds have attracted substantial attention due to their ubiquity in the cores of natural products and bioactive molecules. Among the various synthetic approaches to nitrogenous frameworks, catalytic asymmetric 1,3-dipolar cycloadditions are one of the most attractive methods because of their powerful ability to rapidly construct various chiral N-heterocycles. In particular, N-metallated azomethine ylides, common and readily available 1,3-dipoles, have been extensively applied in dipolar cycloaddition reactions. Despite the fact that asymmetric transformations of azomethine ylides have been investigated for decades, most of the efforts have been directed toward the preparation of pyrrolidines using glycinate-derived α-unsubstituted aldimine esters as the precursors of the azomethine ylides. While α-substituted azomethine ylides derived from amino esters other than glycinate have seldom been harnessed, the construction of non-five-membered chiral N-heterocycles via 1,3-dipolar cycloadditions remains underexplored. In addition, the asymmetric α-functionalization of aldimine esters to prepare acyclic nitrogenous compounds such as α-amino acids, in which an in situ-generated N-metallated azomethine ylide serves as the nucleophile, has not been sufficiently described.In this Account, we mainly discuss the achievements we have made in the past decade toward broadening the applications of N-metallated azomethine ylides for the preparation of nitrogen-containing compounds. We began our investigation with the design and synthesis of a new type of chiral ligand, TF-BiphamPhos, which not only coordinates with Lewis acids to activate dipolar species but also serves as an H-bond donor to increase the reactivity of dipolarophiles with significantly enhanced stereochemical control. Using the Cu(I) or Ag(I)/TF-BiphamPhos complex as the catalyst, we achieved highly stereoselective (3+2) cycloadditions of glycinate and non-glycinate-derived azomethine ylides with diverse dipolarophiles, producing a variety of enantioenriched pyrrolidines with multiple stereocenters in a single step. To further expand the synthetic utility of N-metallated azomethine ylides, we successfully developed higher order cycloadditions with fulvenes, tropone, 2-acyl cycloheptatrienes, and pyrazolidinium ylides serving as the reaction partner, and this reaction provides straightforward access to enantioenriched fused piperidines, bridged azabicyclic frameworks, and triazines via (3+6)- and (3+3)-type cycloadditions. Using N-metallated azomethine ylides as the nucleophile, we realized Cu(I)-catalyzed asymmetric 1,4-Michael additions with α,β-unsaturated bisphosphates/Morita-Baylis-Hillman products, furnishing an array of structurally diverse unnatural α-amino acids. Based on the strategy of synergistic activation, we achieved highly efficient dual Cu/Pd and Cu/Ir catalysis for the α-functionalization of aldimine esters via the asymmetric allylic/allenylic alkylation of N-metallated azomethine ylides. Notably, Cu/Ir catalysis allowed the stereodivergent synthesis of α,α-disubstituted α-amino acids via a branched allylic alkylation reaction, in which the two distinct chiral metal catalysts independently have full stereochemical control over the corresponding nucleophile and electrophile. Furthermore, an expedient and stereodivergent preparation of biologically important tetrahydro-γ-carbolines was realized through a Cu/Ir-catalyzed cascade allylation/iso-Pictet-Spengler cyclization. In addition, when the steric congestion in the allylation intermediates was increased, the combined Cu/Ir catalysts provided an asymmetric cascade allylation/2-aza-Cope rearrangement, producing various optically active homoallylic amines with impressive results.
中文翻译:

N-金属化甲亚胺叶立德的催化不对称反应。
概述由于光活性含氮化合物在天然产物和生物活性分子的核心中普遍存在,因此引起了广泛的关注。在各种合成含氮骨架的方法中,催化不对称1,3-偶极环加成是最吸引人的方法之一,因为它们具有快速构建各种手性N-杂环的强大能力。特别地,N-金属化的偶氮甲亚胺,是常见的且容易获得的1,3-偶极,已广泛用于偶极环加成反应中。尽管已经研究了数十年的甲亚胺基团不对称转化事实,但是大多数努力都针对使用甘氨酸衍生的α-未取代的亚胺基亚胺作为偶氮基亚胺盐的前体来制备吡咯烷。尽管很少利用衍生自甘氨酸以外的氨基酯的α-取代的甲亚胺基化物,但通过1,3-偶极环加成反应的非五元手性N-杂环的构造仍未得到开发。另外,还没有充分描述醛亚胺酯的不对称α-官能化以制备无环含氮化合物,例如α-氨基酸,其中原位产生的N-金属化的偶氮甲亚胺叶立德用作亲核试剂。我们主要讨论在过去十年中在扩大N-金属化的甲亚胺基化物在制备含氮化合物中的应用方面所取得的成就。我们从设计和合成新型手性配体TF-BiphamPhos开始研究,它不仅与路易斯酸配位以激活偶极物种,而且还作为氢键供体,通过显着增强的立体化学控制来提高偶极亲和性的反应性。使用Cu(I)或Ag(I)/ TF-BiphamPhos络合物作为催化剂,我们实现了甘氨酸和非甘氨酸衍生的偶氮甲碱与各种双极性亲和剂的高度立体选择性(3 + 2)环加成反应,产生了多种对映体富集的吡咯烷在一个步骤中具有多个立体中心。为了进一步扩大N-金属化的甲亚胺基团的合成效用,我们成功地开发了以富烯,托普酮,2-酰基环庚烯和吡唑烷鎓基团为反应伙伴的高阶环加成反应,该反应可直接获得对映体富集的稠合哌啶,通过(3 + 6)-和(3 + 3)型环加成键连接桥接的双氮杂双环骨架和三嗪。使用N-金属化的甲亚胺基团作为亲核试剂,我们实现了Cu(I)与α,β-不饱和双磷酸酯/ Morita-Baylis-Hillman产品的不对称1,4-Michael加成,提供了一系列结构多样的非天然α-氨基酸。基于协同活化的策略,我们通过N-金属化的甲亚胺基团的不对称烯丙基/烯基烷基化,实现了高效的Cu / Pd和Cu / Ir双重催化亚胺酯的α-官能化。值得注意的是,Cu / Ir催化通过支链烯丙基烷基化反应实现了立体异构合成α,α-二取代的α-氨基酸,其中两种不同的手性金属催化剂独立地对相应的亲核试剂和亲电试剂具有完全的立体化学控制。此外,通过Cu / Ir催化的级联烯丙基化/ iso-Pictet-Spengler环化反应,实现了重要的生物重要的四氢-γ-咔啉的立体异构制备。另外,当烯丙基化中间体中的空间拥挤增加时,组合的Cu / Ir催化剂提供了不对称的级联烯丙基化/ 2-氮杂-Cope重排,产生了各种具有光学活性的均烯丙基胺,其结果令人印象深刻。
更新日期:2020-04-22
中文翻译:
N-金属化甲亚胺叶立德的催化不对称反应。
概述由于光活性含氮化合物在天然产物和生物活性分子的核心中普遍存在,因此引起了广泛的关注。在各种合成含氮骨架的方法中,催化不对称1,3-偶极环加成是最吸引人的方法之一,因为它们具有快速构建各种手性N-杂环的强大能力。特别地,N-金属化的偶氮甲亚胺,是常见的且容易获得的1,3-偶极,已广泛用于偶极环加成反应中。尽管已经研究了数十年的甲亚胺基团不对称转化事实,但是大多数努力都针对使用甘氨酸衍生的α-未取代的亚胺基亚胺作为偶氮基亚胺盐的前体来制备吡咯烷。尽管很少利用衍生自甘氨酸以外的氨基酯的α-取代的甲亚胺基化物,但通过1,3-偶极环加成反应的非五元手性N-杂环的构造仍未得到开发。另外,还没有充分描述醛亚胺酯的不对称α-官能化以制备无环含氮化合物,例如α-氨基酸,其中原位产生的N-金属化的偶氮甲亚胺叶立德用作亲核试剂。我们主要讨论在过去十年中在扩大N-金属化的甲亚胺基化物在制备含氮化合物中的应用方面所取得的成就。我们从设计和合成新型手性配体TF-BiphamPhos开始研究,它不仅与路易斯酸配位以激活偶极物种,而且还作为氢键供体,通过显着增强的立体化学控制来提高偶极亲和性的反应性。使用Cu(I)或Ag(I)/ TF-BiphamPhos络合物作为催化剂,我们实现了甘氨酸和非甘氨酸衍生的偶氮甲碱与各种双极性亲和剂的高度立体选择性(3 + 2)环加成反应,产生了多种对映体富集的吡咯烷在一个步骤中具有多个立体中心。为了进一步扩大N-金属化的甲亚胺基团的合成效用,我们成功地开发了以富烯,托普酮,2-酰基环庚烯和吡唑烷鎓基团为反应伙伴的高阶环加成反应,该反应可直接获得对映体富集的稠合哌啶,通过(3 + 6)-和(3 + 3)型环加成键连接桥接的双氮杂双环骨架和三嗪。使用N-金属化的甲亚胺基团作为亲核试剂,我们实现了Cu(I)与α,β-不饱和双磷酸酯/ Morita-Baylis-Hillman产品的不对称1,4-Michael加成,提供了一系列结构多样的非天然α-氨基酸。基于协同活化的策略,我们通过N-金属化的甲亚胺基团的不对称烯丙基/烯基烷基化,实现了高效的Cu / Pd和Cu / Ir双重催化亚胺酯的α-官能化。值得注意的是,Cu / Ir催化通过支链烯丙基烷基化反应实现了立体异构合成α,α-二取代的α-氨基酸,其中两种不同的手性金属催化剂独立地对相应的亲核试剂和亲电试剂具有完全的立体化学控制。此外,通过Cu / Ir催化的级联烯丙基化/ iso-Pictet-Spengler环化反应,实现了重要的生物重要的四氢-γ-咔啉的立体异构制备。另外,当烯丙基化中间体中的空间拥挤增加时,组合的Cu / Ir催化剂提供了不对称的级联烯丙基化/ 2-氮杂-Cope重排,产生了各种具有光学活性的均烯丙基胺,其结果令人印象深刻。








































 京公网安备 11010802027423号
京公网安备 11010802027423号