当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Double hybrid DFT calculations with Slater type orbitals
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-04-16 , DOI: 10.1002/jcc.26209 Arno Förster 1 , Lucas Visscher 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2020-04-16 , DOI: 10.1002/jcc.26209 Arno Förster 1 , Lucas Visscher 1
Affiliation
|
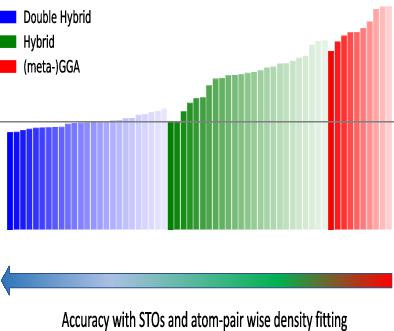
On a comprehensive database with 1,644 datapoints, covering several aspects of main‐group as well as of transition metal chemistry, we assess the performance of 60 density functional approximations (DFA), among them 36 double hybrids (DH). All calculations are performed using a Slater type orbital (STO) basis set of triple‐ζ (TZ) quality and the highly efficient pair atomic resolution of the identity approach for the exchange‐ and Coulomb‐term of the KS matrix (PARI‐K and PARI‐J, respectively) and for the evaluation of the MP2 energy correction (PARI‐MP2). Employing the quadratic scaling SOS‐AO‐PARI‐MP2 algorithm, DHs based on the spin‐opposite‐scaled (SOS) MP2 approximation are benchmarked against a database of large molecules. We evaluate the accuracy of STO/PARI calculations for B3LYP as well as for the DH B2GP‐PLYP and show that the combined basis set and PARI‐error is comparable to the one obtained using the well‐known def2‐TZVPP Gaussian‐type basis set in conjunction with global density fitting. While quadruple‐ζ (QZ) calculations are currently not feasible for PARI‐MP2 due to numerical issues, we show that, on the TZ level, Jacob's ladder for classifying DFAs is reproduced. However, while the best DHs are more accurate than the best hybrids, the improvements are less pronounced than the ones commonly found on the QZ level. For conformers of organic molecules and noncovalent interactions where very high accuracy is required for qualitatively correct results, DHs provide only small improvements over hybrids, while they still excel in thermochemistry, kinetics, transition metal chemistry and the description of strained organic systems.
中文翻译:

使用 Slater 型轨道的双混合 DFT 计算
在包含 1,644 个数据点(涵盖主族以及过渡金属化学的多个方面)的综合数据库上,我们评估了 60 个密度泛函近似 (DFA) 的性能,其中包括 36 个双杂化 (DH)。所有计算均使用三重 z (TZ) 质量的斯莱特型轨道 (STO) 基组和 KS 矩阵交换项和库仑项的恒等方法的高效对原子分辨率(PARI-K 和PARI-J,分别)和 MP2 能量校正的评估(PARI-MP2)。采用二次标度 SOS-AO-PARI-MP2 算法,基于自旋相反标度 (SOS) MP2 近似的 DH 以大分子数据库为基准。我们评估了 B3LYP 以及 DH B2GP-PLYP 的 STO/PARI 计算的准确性,并表明组合基组和 PARI 误差与使用众所周知的 def2-TZVPP 高斯型基组获得的结果相当与全局密度拟合相结合。虽然由于数值问题,四重 z (QZ) 计算目前对于 PARI-MP2 不可行,但我们表明,在 TZ 级别上,再现了用于分类 DFA 的雅各布阶梯。然而,虽然最好的 DH 比最好的混合模型更准确,但其改进不如 QZ 级别上常见的改进那么明显。对于有机分子的构象异构体和非共价相互作用,需要非常高的准确度才能获得正确的定性结果,DH 仅比杂化物提供了很小的改进,但它们在热化学、动力学、过渡金属化学和应变有机系统的描述方面仍然表现出色。
更新日期:2020-04-16
中文翻译:
使用 Slater 型轨道的双混合 DFT 计算
在包含 1,644 个数据点(涵盖主族以及过渡金属化学的多个方面)的综合数据库上,我们评估了 60 个密度泛函近似 (DFA) 的性能,其中包括 36 个双杂化 (DH)。所有计算均使用三重 z (TZ) 质量的斯莱特型轨道 (STO) 基组和 KS 矩阵交换项和库仑项的恒等方法的高效对原子分辨率(PARI-K 和PARI-J,分别)和 MP2 能量校正的评估(PARI-MP2)。采用二次标度 SOS-AO-PARI-MP2 算法,基于自旋相反标度 (SOS) MP2 近似的 DH 以大分子数据库为基准。我们评估了 B3LYP 以及 DH B2GP-PLYP 的 STO/PARI 计算的准确性,并表明组合基组和 PARI 误差与使用众所周知的 def2-TZVPP 高斯型基组获得的结果相当与全局密度拟合相结合。虽然由于数值问题,四重 z (QZ) 计算目前对于 PARI-MP2 不可行,但我们表明,在 TZ 级别上,再现了用于分类 DFA 的雅各布阶梯。然而,虽然最好的 DH 比最好的混合模型更准确,但其改进不如 QZ 级别上常见的改进那么明显。对于有机分子的构象异构体和非共价相互作用,需要非常高的准确度才能获得正确的定性结果,DH 仅比杂化物提供了很小的改进,但它们在热化学、动力学、过渡金属化学和应变有机系统的描述方面仍然表现出色。










































 京公网安备 11010802027423号
京公网安备 11010802027423号