当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts.
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2020-04-14 , DOI: 10.1021/acs.accounts.0c00090 Xiao-Ye Yu 1 , Quan-Qing Zhao 1 , Jun Chen 1 , Wen-Jing Xiao 1 , Jia-Rong Chen 1
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2020-04-14 , DOI: 10.1021/acs.accounts.0c00090 Xiao-Ye Yu 1 , Quan-Qing Zhao 1 , Jun Chen 1 , Wen-Jing Xiao 1 , Jia-Rong Chen 1
Affiliation
|
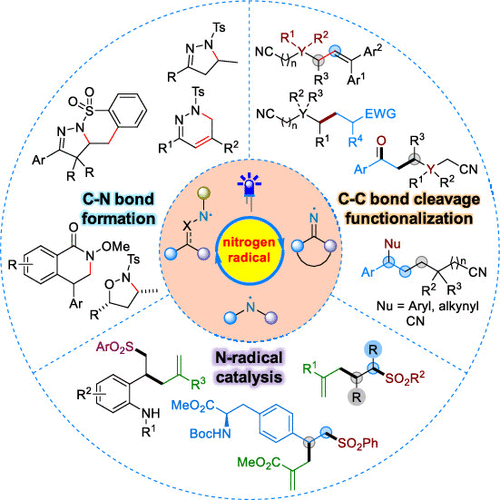
ConspectusNitrogen-centered radicals (NCRs) are a versatile class of highly reactive species that have a longer history than the classical carbon-based radicals in synthetic chemistry. Depending on the N-hybridization and substitution patterns, NCRs can serve as electrophiles or nucleophiles to undergo various radical transformations. Despite their power, progress in nitrogen-radical chemistry is still slow compared with the popularity of carbon radicals, and their considerable synthetic potential has been largely underexplored, which is, as concluded by Zard, mainly hampered by "a dearth of convenient access to these species and a lack of awareness pertaining to their reactivity".Over the past decade, visible-light photoredox catalysis has been established as a powerful toolbox that synthetic chemists can use to generate a diverse range of radical intermediates from native organic functional groups via a single electron transfer process or energy transfer under mild reaction conditions. This catalytic strategy typically obviates the need for external stoichiometric activation reagents or toxic initiators and often enables traditionally inaccessible ionic chemical reactions. On the basis of our long-standing interest in nitrogen chemistry and catalysis, we have emphasized the use of visible-light photoredox catalysis as a tactic to discover and develop novel methods for generating NCRs in a controlled fashion and synthetic applications. In this Account, we describe our recent advances in the development of visible-light-driven photoredox-catalyzed generation of NCRs and their synthetic applications.Inspired by the natural biological proton-coupled electron transfer (PCET) process, we first developed a strategy of visible-light-driven photoredox-catalyzed oxidative deprotonation electron transfer to activate the N-H bonds of hydrazones, benzamides, and sulfonamides to give the corresponding NCRs under mild reaction conditions. With these reactive species, we then achieved a range of 5-exo and 6-endo radical cyclizations as well as cascade reactions in a highly regioselective manner, providing access to a variety of potentially useful nitrogen heterocycles. To further expand the repertoire of possible reactions of NCRs, we also revealed that iminyl radicals, derived from O-acyl cycloalkanone oxime esters, can undergo facile ring-opening C-C bond cleavage to give cyanoalkyl radicals. These newly formed radical species can further undergo a variety of C-C bond-forming reactions to allow the synthesis of diverse distally functionalized alkyl nitriles. Stimulated by these studies, we further developed a wide variety of visible-light-driven copper-catalyzed radical cross-coupling reactions of cyanoalkyl radicals. Because of their inherent highly reactive and transient properties, the strategy of heteroatom-centered radical catalysis is still largely underexplored in organic synthesis. Building on our understanding of the fundamental chemistry of NCRs, we also developed for the first time the concept of NCR covalent catalysis, which involves the use of in situ-photogenerated NCRs to activate allyl sulfones, vinylcyclopropanes, and N-tosyl vinylaziridines. This catalytic strategy has thus enabled efficient difunctionalization of various alkenes and late-stage modification of complex biologically active molecules.In this Account, we describe a panoramic picture of our recent contributions since 2014 to the development and application of the visible-light-driven photoredox systems in the field of NCR chemistry. These studies provide not only efficient methods for the synthesis of functionally rich molecules but also some insight into the exploration of new reactivity or reaction modes of NCRs.
中文翻译:

当光遇到以氮为中心的自由基时:从试剂到催化剂。
概述以氮为中心的自由基(NCR)是一类多用途的高反应性物种,其历史比合成化学中的经典碳基自由基更长。取决于N-杂交和取代模式,NCR可以用作亲电子试剂或亲核试剂以进行各种自由基转化。尽管具有强大的功能,但与碳自由基的普及相比,氮自由基化学的进展仍然很缓慢,并且其相当大的合成潜力尚未得到充分开发,正如扎德(Zard)总结的那样,主要是因为“缺乏方便地获得这些自由基的能力”物种,缺乏对它们的反应性的认识”。在过去十年中,可见光光氧化还原催化已被确立为一个强大的工具箱,合成化学家可以使用它通过单电子转移过程或在温和的反应条件下进行的能量转移,从天然有机官能团生成多种自由基中间体。这种催化策略通常消除了对外部化学计量活化试剂或有毒引发剂的需求,并且通常使传统上无法实现的离子化学反应成为可能。基于我们长期以来对氮化学和催化的兴趣,我们强调了使用可见光光氧化还原催化作为发现和开发以受控方式和合成应用生成NCR的新方法的策略。在这个帐户中,我们描述了在可见光驱动的光氧化还原催化的NCR的开发及其合成应用方面的最新进展。受自然生物质子耦合电子转移(PCET)过程的启发,我们首先开发了可见光策略。驱动光氧化还原催化的氧化去质子电子转移,以活化,苯甲酰胺和磺酰胺的NH键,在温和的反应条件下得到相应的NCR。然后,利用这些反应性物种,我们以高度区域选择性的方式实现了一系列5-exo和6-endo自由基环化以及级联反应,从而提供了各种潜在有用的氮杂环的途径。为了进一步扩大NCR可能反应的范围,我们还揭示了亚氨基,衍生自O-酰基环烷酮肟酯的酯,可进行容易的开环CC键裂解,得到氰基烷基。这些新形成的自由基物质可进一步经历各种CC键形成反应,以合成各种远端官能化的烷基腈。在这些研究的刺激下,我们进一步开发了各种可见光驱动的铜催化的氰基烷基自由基交叉偶联反应。由于其固有的高反应性和瞬态特性,在有机合成中仍未充分研究以杂原子为中心的自由基催化策略。基于对NCR基本化学的理解,我们还首次开发了NCR共价催化的概念,涉及使用原位光生NCR活化烯丙基砜,乙烯基环丙烷和N-甲苯磺酰基乙烯基氮丙啶。因此,这种催化策略使各种烯烃能够高效地进行双官能化,并对复杂的生物活性分子进行后期修饰。在本报告中,我们描述了自2014年以来我们对可见光驱动的光氧化还原的发展和应用所做的最新贡献的全景图。 NCR化学领域的系统。这些研究不仅为合成功能丰富的分子提供了有效的方法,而且为探索NCR的新反应性或反应模式提供了一些见识。我们将概述自2014年以来我们对NCR化学领域中可见光驱动的光氧化还原系统的开发和应用所做的最新贡献。这些研究不仅为合成功能丰富的分子提供了有效的方法,而且为探索NCR的新反应性或反应模式提供了一些见识。我们将概述自2014年以来我们对NCR化学领域中可见光驱动的光氧化还原系统的开发和应用所做的最新贡献。这些研究不仅为合成功能丰富的分子提供了有效的方法,而且为探索NCR的新反应性或反应模式提供了一些见识。
更新日期:2020-04-14
中文翻译:
当光遇到以氮为中心的自由基时:从试剂到催化剂。
概述以氮为中心的自由基(NCR)是一类多用途的高反应性物种,其历史比合成化学中的经典碳基自由基更长。取决于N-杂交和取代模式,NCR可以用作亲电子试剂或亲核试剂以进行各种自由基转化。尽管具有强大的功能,但与碳自由基的普及相比,氮自由基化学的进展仍然很缓慢,并且其相当大的合成潜力尚未得到充分开发,正如扎德(Zard)总结的那样,主要是因为“缺乏方便地获得这些自由基的能力”物种,缺乏对它们的反应性的认识”。在过去十年中,可见光光氧化还原催化已被确立为一个强大的工具箱,合成化学家可以使用它通过单电子转移过程或在温和的反应条件下进行的能量转移,从天然有机官能团生成多种自由基中间体。这种催化策略通常消除了对外部化学计量活化试剂或有毒引发剂的需求,并且通常使传统上无法实现的离子化学反应成为可能。基于我们长期以来对氮化学和催化的兴趣,我们强调了使用可见光光氧化还原催化作为发现和开发以受控方式和合成应用生成NCR的新方法的策略。在这个帐户中,我们描述了在可见光驱动的光氧化还原催化的NCR的开发及其合成应用方面的最新进展。受自然生物质子耦合电子转移(PCET)过程的启发,我们首先开发了可见光策略。驱动光氧化还原催化的氧化去质子电子转移,以活化,苯甲酰胺和磺酰胺的NH键,在温和的反应条件下得到相应的NCR。然后,利用这些反应性物种,我们以高度区域选择性的方式实现了一系列5-exo和6-endo自由基环化以及级联反应,从而提供了各种潜在有用的氮杂环的途径。为了进一步扩大NCR可能反应的范围,我们还揭示了亚氨基,衍生自O-酰基环烷酮肟酯的酯,可进行容易的开环CC键裂解,得到氰基烷基。这些新形成的自由基物质可进一步经历各种CC键形成反应,以合成各种远端官能化的烷基腈。在这些研究的刺激下,我们进一步开发了各种可见光驱动的铜催化的氰基烷基自由基交叉偶联反应。由于其固有的高反应性和瞬态特性,在有机合成中仍未充分研究以杂原子为中心的自由基催化策略。基于对NCR基本化学的理解,我们还首次开发了NCR共价催化的概念,涉及使用原位光生NCR活化烯丙基砜,乙烯基环丙烷和N-甲苯磺酰基乙烯基氮丙啶。因此,这种催化策略使各种烯烃能够高效地进行双官能化,并对复杂的生物活性分子进行后期修饰。在本报告中,我们描述了自2014年以来我们对可见光驱动的光氧化还原的发展和应用所做的最新贡献的全景图。 NCR化学领域的系统。这些研究不仅为合成功能丰富的分子提供了有效的方法,而且为探索NCR的新反应性或反应模式提供了一些见识。我们将概述自2014年以来我们对NCR化学领域中可见光驱动的光氧化还原系统的开发和应用所做的最新贡献。这些研究不仅为合成功能丰富的分子提供了有效的方法,而且为探索NCR的新反应性或反应模式提供了一些见识。我们将概述自2014年以来我们对NCR化学领域中可见光驱动的光氧化还原系统的开发和应用所做的最新贡献。这些研究不仅为合成功能丰富的分子提供了有效的方法,而且为探索NCR的新反应性或反应模式提供了一些见识。










































 京公网安备 11010802027423号
京公网安备 11010802027423号