当前位置:
X-MOL 学术
›
J. Rare Earths
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First-principles study of electronic structure and optical properties of Er:Lu2O3
Journal of Rare Earths ( IF 5.2 ) Pub Date : 2020-03-01 , DOI: 10.1016/j.jre.2020.03.001 Xian Zhang , Honglei Zhao , Sen Gao , Qingfeng Zeng
Journal of Rare Earths ( IF 5.2 ) Pub Date : 2020-03-01 , DOI: 10.1016/j.jre.2020.03.001 Xian Zhang , Honglei Zhao , Sen Gao , Qingfeng Zeng
|
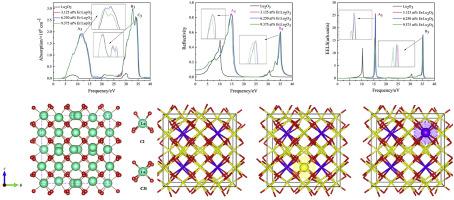
Abstract In the present computational study, we found that Er:Lu2O3 materials have promise for application in laser applications. The crystal structure and the electronic and optical properties of Er:Lu2O3 materials were studied using first-principle calculations under the framework of density functional theory. Based on the experimental and calculated results, the structure of Lu2O3 was established. The calculated results show that doping by Er3+ can effectively improve its absorption coefficient in the ultraviolet region and improve the static dielectric constant of Lu2O3. As the doping concentration of Er3+ increases, the energy of the valence band electrons excited to the conduction band decreases, and the transition is more likely to occur. The absorption coefficient, reflectance, and electron energy loss spectroscopy are bathochromic shifted. The Lu2-xErxO3 (0
中文翻译:

Er:Lu2O3电子结构和光学性质的第一性原理研究
摘要 在目前的计算研究中,我们发现 Er:Lu2O3 材料在激光应用中具有应用前景。在密度泛函理论的框架下,使用第一性原理计算研究了 Er:Lu2O3 材料的晶体结构以及电子和光学性质。根据实验和计算结果,建立了Lu2O3的结构。计算结果表明,Er3+掺杂可以有效提高其在紫外区的吸收系数,提高Lu2O3的静态介电常数。随着Er3+掺杂浓度的增加,激发到导带的价带电子的能量降低,更容易发生跃迁。吸收系数、反射率和电子能量损失光谱是红移的。
更新日期:2020-03-01
中文翻译:
Er:Lu2O3电子结构和光学性质的第一性原理研究
摘要 在目前的计算研究中,我们发现 Er:Lu2O3 材料在激光应用中具有应用前景。在密度泛函理论的框架下,使用第一性原理计算研究了 Er:Lu2O3 材料的晶体结构以及电子和光学性质。根据实验和计算结果,建立了Lu2O3的结构。计算结果表明,Er3+掺杂可以有效提高其在紫外区的吸收系数,提高Lu2O3的静态介电常数。随着Er3+掺杂浓度的增加,激发到导带的价带电子的能量降低,更容易发生跃迁。吸收系数、反射率和电子能量损失光谱是红移的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号