Computational and Structural Biotechnology Journal ( IF 4.4 ) Pub Date : 2020-02-20 , DOI: 10.1016/j.csbj.2020.02.007 Debby D Wang 1 , Le Ou-Yang 2 , Haoran Xie 3 , Mengxu Zhu 4 , Hong Yan 4
|
Purpose
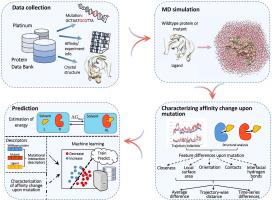
Mutation-induced variation of protein-ligand binding affinity is the key to many genetic diseases and the emergence of drug resistance, and therefore predicting such mutation impacts is of great importance. In this work, we aim to predict the mutation impacts on protein-ligand binding affinity using efficient structure-based, computational methods.
Methods
Relying on consolidated databases of experimentally determined data we characterize the affinity change upon mutation based on a number of local geometrical features and monitor such feature differences upon mutation during molecular dynamics (MD) simulations. The differences are quantified according to average difference, trajectory-wise distance or time-vary differences. Machine-learning methods are employed to predict the mutation impacts using the resulting conventional or time-series features. Predictions based on estimation of energy and based on investigation of molecular descriptors were conducted as benchmarks.
Results
Our method (machine-learning techniques using time-series features) outperformed the benchmark methods, especially in terms of the balanced F1 score. Particularly, deep-learning models led to the best prediction performance with distinct improvements in balanced F1 score and a sustained accuracy.
Conclusion
Our work highlights the effectiveness of the characterization of affinity change upon mutations. Furthermore, deep-learning techniques are well designed for handling the extracted time-series features. This study can lead to a deeper understanding of mutation-induced diseases and resistance, and further guide the development of innovative drug design.
中文翻译:
基于分子动力学模拟和机器学习方法预测突变对蛋白质-配体结合亲和力的影响
目的
突变引起的蛋白质-配体结合亲和力的变化是许多遗传疾病和耐药性出现的关键,因此预测这种突变影响非常重要。在这项工作中,我们旨在使用有效的基于结构的计算方法来预测突变对蛋白质-配体结合亲和力的影响。
方法
依靠实验确定数据的综合数据库,我们基于许多局部几何特征来表征突变后的亲和力变化,并在分子动力学(MD)模拟期间监测突变后的这种特征差异。根据平均差异、轨迹距离或时变差异对差异进行量化。机器学习方法被用来使用产生的常规或时间序列特征来预测突变影响。基于能量估计和分子描述符研究的预测被作为基准进行。
结果
我们的方法(使用时间序列特征的机器学习技术)优于基准方法,尤其是在平衡 F1 分数方面。特别是,深度学习模型带来了最佳的预测性能,在平衡 F1 分数和持续准确性方面有显着提高。
结论
我们的工作强调了突变后亲和力变化表征的有效性。此外,深度学习技术经过精心设计,可以处理提取的时间序列特征。该研究可以加深对突变引起的疾病和耐药性的认识,并进一步指导创新药物设计的发展。










































 京公网安备 11010802027423号
京公网安备 11010802027423号