Computational and Structural Biotechnology Journal ( IF 4.4 ) Pub Date : 2020-03-06 , DOI: 10.1016/j.csbj.2020.02.021 Chi-Wei Chen , Meng-Han Lin , Chi-Chou Liao , Hsung-Pin Chang , Yen-Wei Chu
|
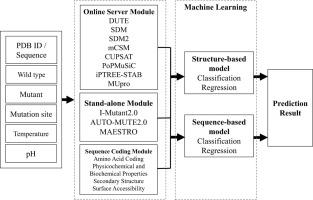
Protein mutations can lead to structural changes that affect protein function and result in disease occurrence. In protein engineering, drug design or and optimization industries, mutations are often used to improve protein stability or to change protein properties while maintaining stability. To provide possible candidates for novel protein design, several computational tools for predicting protein stability changes have been developed. Although many prediction tools are available, each tool employs different algorithms and features. This can produce conflicting prediction results that make it difficult for users to decide upon the correct protein design. Therefore, this study proposes an integrated prediction tool, iStable 2.0, which integrates 11 sequence-based and structure-based prediction tools by machine learning and adds protein sequence information as features. Three coding modules are designed for the system, an Online Server Module, a Stand-alone Module and a Sequence Coding Module, to improve the prediction performance of the previous version of the system. The final integrated structure-based classification model has a higher Matthews correlation coefficient than that of the single prediction tool (0.708 vs 0.547, respectively), and the Pearson correlation coefficient of the regression model likewise improves from 0.669 to 0.714. The sequence-based model not only successfully integrates off-the-shelf predictors but also improves the Matthews correlation coefficient of the best single prediction tool by at least 0.161, which is better than the individual structure-based prediction tools. In addition, both the Sequence Coding Module and the Stand-alone Module maintain performance with only a 5% decrease of the Matthews correlation coefficient when the integrated online tools are unavailable. iStable 2.0 is available at http://ncblab.nchu.edu.tw/iStable2.
中文翻译:
iStable 2.0:通过集成各种特性模块来预测蛋白质的热稳定性变化
蛋白质突变可导致影响蛋白质功能的结构变化并导致疾病发生。在蛋白质工程,药物设计或优化行业中,突变通常用于改善蛋白质稳定性或更改蛋白质特性,同时保持稳定性。为了提供新颖蛋白质设计的可能候选者,已经开发了几种用于预测蛋白质稳定性变化的计算工具。尽管有许多预测工具可用,但是每种工具都采用不同的算法和功能。这可能会产生相互矛盾的预测结果,使用户难以决定正确的蛋白质设计。因此,本研究提出了一种集成的预测工具iStable 2.0,它通过机器学习集成了11种基于序列和基于结构的预测工具,并添加了蛋白质序列信息作为特征。为系统设计了三个编码模块,一个在线服务器模块,一个独立模块和一个序列编码模块,以提高系统先前版本的预测性能。最终基于集成结构的分类模型比单个预测工具具有更高的Matthews相关系数(分别为0.708和0.547),回归模型的Pearson相关系数也从0.669改善到0.714。基于序列的模型不仅成功集成了现成的预测变量,而且还将最佳单一预测工具的Matthews相关系数提高了至少0.161,这比单个基于结构的预测工具要好。此外,当集成的在线工具不可用时,序列编码模块和独立模块都可以保持性能,并且仅使Matthews相关系数降低5%。iStable 2.0可从http://ncblab.nchu.edu.tw/iStable2获得。










































 京公网安备 11010802027423号
京公网安备 11010802027423号