Nature Machine Intelligence ( IF 18.8 ) Pub Date : 2020-04-06 , DOI: 10.1038/s42256-020-0167-4 Ruibang Luo , Chak-Lim Wong , Yat-Sing Wong , Chi-Ian Tang , Chi-Man Liu , Chi-Ming Leung , Tak-Wah Lam
|
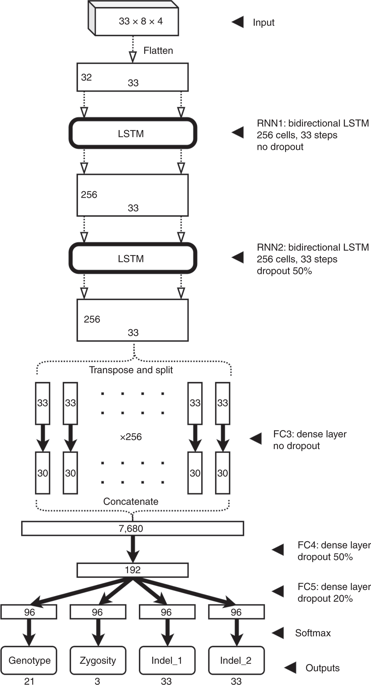
Single-molecule sequencing technologies have emerged in recent years and revolutionized structural variant calling, complex genome assembly and epigenetic mark detection. However, the lack of a highly accurate small variant caller has limited these technologies from being more widely used. Here, we present Clair, the successor to Clairvoyante, a program for fast and accurate germline small variant calling, using single-molecule sequencing data. For Oxford Nanopore Technology data, Clair achieves better precision, recall and speed than several competing programs, including Clairvoyante, Longshot and Medaka. Through studying the missed variants and benchmarking intentionally overfitted models, we found that Clair may be approaching the limit of possible accuracy for germline small variant calling using pileup data and deep neural networks. Clair requires only a conventional central processing unit (CPU) for variant calling and is an open-source project available at https://github.com/HKU-BAL/Clair.
A preprint version of the article is available at bioRxiv.中文翻译:
探索在堆积数据上使用深度神经网络进行种系变异调用的限制
近年来,单分子测序技术应运而生,彻底改变了结构变异调用,复杂的基因组装配和表观遗传标记检测。但是,由于缺少高度准确的小型变体调用程序,因此无法广泛使用这些技术。在这里,我们介绍Clair,它是Clairvoyante的后继者,Clairvoyante是使用单分子测序数据进行快速准确的种系小变异调用的程序。对于牛津纳米孔技术的数据,Clair的精度,召回率和速度都比包括Clairvoyante,Longshot和Medaka在内的多个竞争程序要好。通过研究遗漏的变体和对有意过拟合的模型进行基准测试,我们发现Clair可能正在利用堆积数据和深度神经网络接近种系小变体调用的可能准确性极限。
文章的预印本可从bioRxiv获得。









































 京公网安备 11010802027423号
京公网安备 11010802027423号