当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Chemical Shift Tensors of Cimetidine Form A Modeled with Density Functional Theory Calculations: Implications for NMR Crystallography.
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2020-04-14 , DOI: 10.1021/acs.jpca.0c00421 Sean T Holmes 1 , Olivia G Engl 2 , Matthew N Srnec 3 , Jeffry D Madura 4 , Rosalynn Quiñones 5 , James K Harper 6 , Robert W Schurko 1 , Robbie J Iuliucci 2
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2020-04-14 , DOI: 10.1021/acs.jpca.0c00421 Sean T Holmes 1 , Olivia G Engl 2 , Matthew N Srnec 3 , Jeffry D Madura 4 , Rosalynn Quiñones 5 , James K Harper 6 , Robert W Schurko 1 , Robbie J Iuliucci 2
Affiliation
|
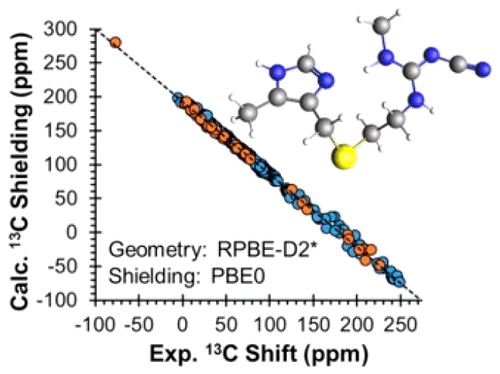
The principal components of the 13C chemical shift tensors for the ten crystallographically distinct carbon atoms of the active pharmaceutical ingredient cimetidine Form A have been measured using the FIREMAT technique. Density functional theory (DFT) calculations of 13C and 15N magnetic shielding tensors are used to assign the 13C and 15N peaks. DFT calculations were performed on cimetidine and a training set of organic crystals using both plane-wave and cluster-based approaches. The former set of calculations allowed several structural refinement strategies to be employed, including calculations utilizing a dispersion-corrected force field that was parametrized using 13C and 15N magnetic shielding tensors. The latter set of calculations featured the use of resource-intensive hybrid-DFT methods for the calculation of magnetic shielding tensors. Calculations on structures refined using the new force-field correction result in improved values of 15N magnetic shielding tensors (as gauged by agreement with experimental chemical shift tensors), although little improvement is seen in the prediction of 13C shielding tensors. Calculations of 13C and 15N magnetic shielding tensors using hybrid functionals show better agreement with experimental values in comparison to those using GGA functionals, independent of the method of structural refinement; the shielding of carbon atoms bonded to nitrogen are especially improved using hybrid DFT methods.
中文翻译:

西咪替丁A型的化学位移张量以密度泛函理论计算为模型:对NMR晶体学的启示。
活性药物成分西咪替丁晶型A的十个晶体学上不同的碳原子的13C化学位移张量的主要成分已使用FIREMAT技术进行了测量。使用13C和15N磁屏蔽张量的密度泛函理论(DFT)计算来指定13C和15N峰值。使用平面波和基于簇的方法对西咪替丁和一组有机晶体进行DFT计算。前一组计算允许采用几种结构改进策略,包括利用使用13C和15N磁屏蔽张量参数化的色散校正力场进行的计算。后一组计算的特点是使用资源密集型混合DFT方法计算磁屏蔽张量。尽管使用13C屏蔽张量的预测几乎没有改善,但使用新的力场校正对结构进行的计算可以提高15N磁屏蔽张量的值(通过与实验化学位移张量的协议进行测量)。与使用GGA功能的计算相比,使用混合功能的13C和15N磁屏蔽张量的计算显示出与实验值更好的一致性,而与结构改进方法无关。杂化DFT方法可以改善与氮原子键合的碳原子的屏蔽。尽管在13C屏蔽张量的预测中看不到什么改善。与使用GGA功能的计算相比,使用混合功能的13C和15N磁屏蔽张量的计算显示出与实验值更好的一致性,而与结构改进方法无关。杂化DFT方法可以改善与氮原子键合的碳原子的屏蔽。尽管在13C屏蔽张量的预测中看不到什么改善。与使用GGA功能的结果相比,使用混合功能的13C和15N磁屏蔽张量的计算显示出与实验值更好的一致性,而与结构改进方法无关。杂化DFT方法可以改善与氮原子键合的碳原子的屏蔽。
更新日期:2020-04-24
中文翻译:
西咪替丁A型的化学位移张量以密度泛函理论计算为模型:对NMR晶体学的启示。
活性药物成分西咪替丁晶型A的十个晶体学上不同的碳原子的13C化学位移张量的主要成分已使用FIREMAT技术进行了测量。使用13C和15N磁屏蔽张量的密度泛函理论(DFT)计算来指定13C和15N峰值。使用平面波和基于簇的方法对西咪替丁和一组有机晶体进行DFT计算。前一组计算允许采用几种结构改进策略,包括利用使用13C和15N磁屏蔽张量参数化的色散校正力场进行的计算。后一组计算的特点是使用资源密集型混合DFT方法计算磁屏蔽张量。尽管使用13C屏蔽张量的预测几乎没有改善,但使用新的力场校正对结构进行的计算可以提高15N磁屏蔽张量的值(通过与实验化学位移张量的协议进行测量)。与使用GGA功能的计算相比,使用混合功能的13C和15N磁屏蔽张量的计算显示出与实验值更好的一致性,而与结构改进方法无关。杂化DFT方法可以改善与氮原子键合的碳原子的屏蔽。尽管在13C屏蔽张量的预测中看不到什么改善。与使用GGA功能的计算相比,使用混合功能的13C和15N磁屏蔽张量的计算显示出与实验值更好的一致性,而与结构改进方法无关。杂化DFT方法可以改善与氮原子键合的碳原子的屏蔽。尽管在13C屏蔽张量的预测中看不到什么改善。与使用GGA功能的结果相比,使用混合功能的13C和15N磁屏蔽张量的计算显示出与实验值更好的一致性,而与结构改进方法无关。杂化DFT方法可以改善与氮原子键合的碳原子的屏蔽。


























 京公网安备 11010802027423号
京公网安备 11010802027423号