当前位置:
X-MOL 学术
›
ACS Appl. Energy Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Unveiling the Role of Hydroxyl Architecture on Polysulfide Trapping for High-Performance Lithium–Sulfur Batteries
ACS Applied Energy Materials ( IF 5.4 ) Pub Date : 2020-03-31 00:00:00 , DOI: 10.1021/acsaem.0c00444 Xiaoyan Ren 1 , Qi Sun 1, 2 , Youliang Zhu 3 , Wenbo Sun 1 , Yang Li 1 , Lehui Lu 1, 2
ACS Applied Energy Materials ( IF 5.4 ) Pub Date : 2020-03-31 00:00:00 , DOI: 10.1021/acsaem.0c00444 Xiaoyan Ren 1 , Qi Sun 1, 2 , Youliang Zhu 3 , Wenbo Sun 1 , Yang Li 1 , Lehui Lu 1, 2
Affiliation
|
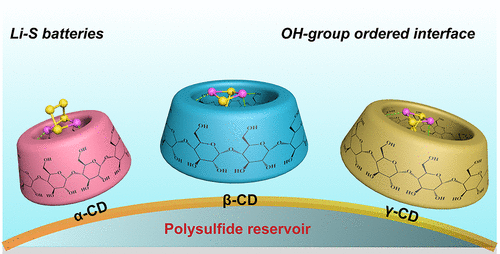
The rapid loss of active sulfur, because of a notorious effect of polysulfide shuttle, leads to a severe capacity fading in lithium–sulfur (Li–S) batteries. Although oxygen doping in cathodic materials is a promising strategy to enhance bonding interactions with lithium polysulfides, the origin of strong interactions remains to be poorly understood, because of a lack of consideration for the spatial arrangement of oxygen atoms affecting the overall performances. Here, we unveil the role of hydroxyl architecture on polysulfide trapping by systematically studying a series of cyclodextrin molecules that serve as a model platform, because of their well-defined structural uniqueness featuring abundant hydroxyl binding sites. We compare their trapping behaviors toward lithium polysulfides and thus correlate performance variations with their structural differences. Experimental findings coupled with computational modeling suggest that the asymmetrical arrangement of primary hydroxyl groups in β-CD determines the highest binding energies, relevant to the optimal capability in constraining polysulfide shuttle, and, in turn, contributes to remarkable improvements in the rate capacity and cycling performance. The identification of the role of hydroxyl architecture provides an atomic-scale explanation for the interaction between OH-group ordered interface and lithium polysulfides, and a new insight for the future design and engineering of interfaces in Li–S batteries.
中文翻译:

揭示羟基结构在高性能锂硫电池多硫化物捕集中的作用
由于多硫化物梭的臭名昭著,活性硫的快速流失导致锂硫(Li–S)电池的严重容量衰减。尽管在阴极材料中掺入氧是增强与多硫化锂之间键合相互作用的一种有前途的策略,但由于缺乏考虑影响整体性能的氧原子的空间排列,因此强相互作用的起源仍然知之甚少。在这里,我们通过系统地研究一系列用作模型平台的环糊精分子,揭示了羟基结构在多硫化物捕获中的作用,因为它们具有丰富的羟基结合位点的结构明确的独特性。我们比较了它们对多硫化锂的捕获行为,因此将性能变化与它们的结构差异相关联。实验发现和计算模型表明,β-CD中伯羟基的不对称排列决定了最高的结合能,这与约束多硫化物穿梭的最佳能力有关,进而有助于显着提高速率能力和循环性能。羟基结构的作用的鉴定为OH-基有序界面与多硫化锂之间的相互作用提供了原子尺度的解释,并为Li-S电池的未来界面设计和工程化提供了新的见解。实验发现和计算模型表明,β-CD中伯羟基的不对称排列决定了最高的结合能,这与约束多硫化物穿梭的最佳能力有关,进而有助于显着提高速率能力和循环性能。羟基结构的作用的鉴定为OH-基有序界面与多硫化锂之间的相互作用提供了原子尺度的解释,并为Li-S电池的未来界面设计和工程化提供了新的见解。实验发现和计算模型表明,β-CD中伯羟基的不对称排列决定了最高的结合能,这与约束多硫化物穿梭的最佳能力有关,进而有助于显着提高速率能力和循环性能。羟基结构的作用的鉴定为OH-基有序界面与多硫化锂之间的相互作用提供了原子尺度的解释,并为Li-S电池的未来界面设计和工程化提供了新的见解。
更新日期:2020-03-31
中文翻译:
揭示羟基结构在高性能锂硫电池多硫化物捕集中的作用
由于多硫化物梭的臭名昭著,活性硫的快速流失导致锂硫(Li–S)电池的严重容量衰减。尽管在阴极材料中掺入氧是增强与多硫化锂之间键合相互作用的一种有前途的策略,但由于缺乏考虑影响整体性能的氧原子的空间排列,因此强相互作用的起源仍然知之甚少。在这里,我们通过系统地研究一系列用作模型平台的环糊精分子,揭示了羟基结构在多硫化物捕获中的作用,因为它们具有丰富的羟基结合位点的结构明确的独特性。我们比较了它们对多硫化锂的捕获行为,因此将性能变化与它们的结构差异相关联。实验发现和计算模型表明,β-CD中伯羟基的不对称排列决定了最高的结合能,这与约束多硫化物穿梭的最佳能力有关,进而有助于显着提高速率能力和循环性能。羟基结构的作用的鉴定为OH-基有序界面与多硫化锂之间的相互作用提供了原子尺度的解释,并为Li-S电池的未来界面设计和工程化提供了新的见解。实验发现和计算模型表明,β-CD中伯羟基的不对称排列决定了最高的结合能,这与约束多硫化物穿梭的最佳能力有关,进而有助于显着提高速率能力和循环性能。羟基结构的作用的鉴定为OH-基有序界面与多硫化锂之间的相互作用提供了原子尺度的解释,并为Li-S电池的未来界面设计和工程化提供了新的见解。实验发现和计算模型表明,β-CD中伯羟基的不对称排列决定了最高的结合能,这与约束多硫化物穿梭的最佳能力有关,进而有助于显着提高速率能力和循环性能。羟基结构的作用的鉴定为OH-基有序界面与多硫化锂之间的相互作用提供了原子尺度的解释,并为Li-S电池的未来界面设计和工程化提供了新的见解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号