当前位置:
X-MOL 学术
›
Appl. Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Metallic two-dimensional C3N allotropes with electron and ion channels for high-performance Li-ion battery anode materials
Applied Surface Science ( IF 6.3 ) Pub Date : 2020-07-01 , DOI: 10.1016/j.apsusc.2020.146254 Gencai Guo , Ruzhi Wang , Siwei Luo , Bangming Ming , Changhao Wang , Ming Zhang , Yuefei Zhang , Hui Yan
Applied Surface Science ( IF 6.3 ) Pub Date : 2020-07-01 , DOI: 10.1016/j.apsusc.2020.146254 Gencai Guo , Ruzhi Wang , Siwei Luo , Bangming Ming , Changhao Wang , Ming Zhang , Yuefei Zhang , Hui Yan
|
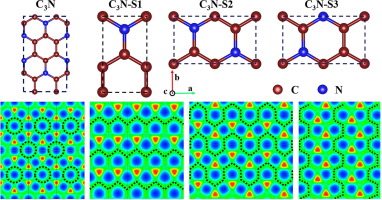
Abstract Two-dimensional monolayer C3N has attracted much attention in many fields, owing to its unique mechanical, electrical and thermal properties and diverse structure. However, the physical origin of these properties lacks a systematic and clear understanding. In this work, three dynamically stable metallic C3N allotropes were computationally investigated. The cohesive energy of all these structures is within 0.09 eV/atom of that of the experimentally reported semi-conductive C3N structure. Density-functional theory calculations showed that the metallicity of two-dimensional C3N can be attributed to the continuity of the carbon chains, which form channels for passage of free electrons. Furthermore, additional calculations showed that the C3N allotropes possess excellent mechanical properties, good electronic conductivity and Li migration capability (wherein the C-C chains provide a good migration channel for lithium ions). These properties make C3N a promising candidate for Li-ion battery anode materials. Our findings suggest a physical mechanistic basis for modulating the electrochemical properties of such materials by controlling the arrangement of atoms. The study also provides new ideas for the modulation of the structure and properties of two-dimensional materials and points out a direction for the ongoing development and application of C-N materials and devices.
中文翻译:

用于高性能锂离子电池负极材料的具有电子和离子通道的金属二维 C3N 同素异形体
摘要 二维单层C3N由于其独特的机械、电学和热学性能以及多样的结构而在许多领域引起了广泛关注。然而,这些性质的物理起源缺乏系统和清晰的认识。在这项工作中,计算研究了三种动态稳定的金属 C3N 同素异形体。所有这些结构的内聚能与实验报告的半导体 C3N 结构的内聚能相差在 0.09 eV/原子以内。密度泛函理论计算表明,二维 C3N 的金属丰度可归因于碳链的连续性,碳链形成了自由电子通过的通道。此外,额外的计算表明 C3N 同素异形体具有优异的机械性能,良好的电子导电性和锂迁移能力(其中 CC 链为锂离子提供了良好的迁移通道)。这些特性使 C3N 成为锂离子电池负极材料的有希望的候选者。我们的研究结果表明,通过控制原子排列来调节此类材料的电化学性质具有物理机制基础。该研究还为二维材料结构和性能的调制提供了新思路,为CN材料和器件的持续发展和应用指明了方向。我们的研究结果表明,通过控制原子排列来调节此类材料的电化学性质具有物理机制基础。该研究还为二维材料结构和性能的调制提供了新思路,为CN材料和器件的持续发展和应用指明了方向。我们的研究结果表明,通过控制原子排列来调节此类材料的电化学性质具有物理机制基础。该研究还为二维材料结构和性能的调制提供了新思路,为CN材料和器件的持续发展和应用指明了方向。
更新日期:2020-07-01
中文翻译:
用于高性能锂离子电池负极材料的具有电子和离子通道的金属二维 C3N 同素异形体
摘要 二维单层C3N由于其独特的机械、电学和热学性能以及多样的结构而在许多领域引起了广泛关注。然而,这些性质的物理起源缺乏系统和清晰的认识。在这项工作中,计算研究了三种动态稳定的金属 C3N 同素异形体。所有这些结构的内聚能与实验报告的半导体 C3N 结构的内聚能相差在 0.09 eV/原子以内。密度泛函理论计算表明,二维 C3N 的金属丰度可归因于碳链的连续性,碳链形成了自由电子通过的通道。此外,额外的计算表明 C3N 同素异形体具有优异的机械性能,良好的电子导电性和锂迁移能力(其中 CC 链为锂离子提供了良好的迁移通道)。这些特性使 C3N 成为锂离子电池负极材料的有希望的候选者。我们的研究结果表明,通过控制原子排列来调节此类材料的电化学性质具有物理机制基础。该研究还为二维材料结构和性能的调制提供了新思路,为CN材料和器件的持续发展和应用指明了方向。我们的研究结果表明,通过控制原子排列来调节此类材料的电化学性质具有物理机制基础。该研究还为二维材料结构和性能的调制提供了新思路,为CN材料和器件的持续发展和应用指明了方向。我们的研究结果表明,通过控制原子排列来调节此类材料的电化学性质具有物理机制基础。该研究还为二维材料结构和性能的调制提供了新思路,为CN材料和器件的持续发展和应用指明了方向。










































 京公网安备 11010802027423号
京公网安备 11010802027423号