当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Treatment of the Coriolis Effect Using Hyperspherical Coordinates, with Application to the Ro-Vibrational Spectrum of Ozone
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-03-31 , DOI: 10.1021/acs.jpca.0c00893 Igor Gayday 1 , Alexander Teplukhin 2 , Brian K. Kendrick 2 , Dmitri Babikov 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2020-03-31 , DOI: 10.1021/acs.jpca.0c00893 Igor Gayday 1 , Alexander Teplukhin 2 , Brian K. Kendrick 2 , Dmitri Babikov 1
Affiliation
|
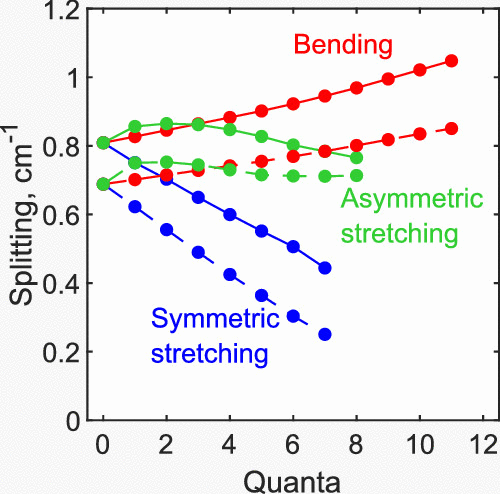
Several alternative methods for the description of the interaction between rotation and vibration are compared and contrasted using hyperspherical coordinates for a triatomic molecule. These methods differ by the choice of the z-axis and by the assumption of a prolate or oblate rotor shape of the molecule. For each case, a block-structure of the rotational–vibrational Hamiltonian matrix is derived and analyzed, and the advantages and disadvantages of each method are made explicit. This theory is then employed to compute ro-vibrational spectra of singly substituted ozone; roughly, 600 vibrational states of 16O18O16O and 16O16O18O isomers combined, with rotational excitations up to J = 5 and both inversion parities (21600 coupled ro-vibrational states in total). Splittings between the states of different parities, so-called K-doublings, are calculated and analyzed. The roles of the asymmetric-top rotor term and the Coriolis coupling term are determined individually, and it is found that they both affect these splittings, but in the opposite directions. Thus, the two effects partially cancel out, and the residual splittings are relatively small. Energies of the ro-vibrational states reported in this work for 16O18O16O and 16O16O18O are in excellent agreement with literature (available for low-vibrational excitation). New data obtained here for the highly excited vibrational states enable the first systematic study of the Coriolis effect in symmetric and asymmetric isotopomers of ozone.
中文翻译:

用超球坐标理论处理科里奥利效应,并应用于臭氧的Ro振动光谱
使用三原子分子的超球坐标比较和对比了描述旋转和振动之间相互作用的几种替代方法。这些方法的不同之处在于对z轴的选择以及对分子的长圆形或扁圆形转子形状的假设。对于每种情况,都导出并分析了旋转振动哈密顿矩阵的块结构,并明确了每种方法的优缺点。然后,利用该理论来计算单取代臭氧的旋转振动光谱。大约有16个O 18 O 16 O和16 O 16 O 18的600个振动状态O异构体结合在一起,具有高达J = 5的旋转激发和两个反位(总共21600个耦合ro振动状态)。计算和分析不同奇偶校验状态之间的分裂,即所谓的K倍增。单独确定不对称顶部转子项和科里奥利耦合项的作用,并且发现它们都影响这些分裂,但方向相反。因此,这两个效果被部分抵消,并且残余裂痕相对较小。在这项工作中报告的16 O 18 O 16 O和16 O 16 O 18的振动状态的能量O与文献(可用于低振动激励)非常一致。此处获得的有关高激发振动态的新数据使我们能够对臭氧对称和非对称异构体中的科里奥利效应进行首次系统研究。
更新日期:2020-03-31
中文翻译:
用超球坐标理论处理科里奥利效应,并应用于臭氧的Ro振动光谱
使用三原子分子的超球坐标比较和对比了描述旋转和振动之间相互作用的几种替代方法。这些方法的不同之处在于对z轴的选择以及对分子的长圆形或扁圆形转子形状的假设。对于每种情况,都导出并分析了旋转振动哈密顿矩阵的块结构,并明确了每种方法的优缺点。然后,利用该理论来计算单取代臭氧的旋转振动光谱。大约有16个O 18 O 16 O和16 O 16 O 18的600个振动状态O异构体结合在一起,具有高达J = 5的旋转激发和两个反位(总共21600个耦合ro振动状态)。计算和分析不同奇偶校验状态之间的分裂,即所谓的K倍增。单独确定不对称顶部转子项和科里奥利耦合项的作用,并且发现它们都影响这些分裂,但方向相反。因此,这两个效果被部分抵消,并且残余裂痕相对较小。在这项工作中报告的16 O 18 O 16 O和16 O 16 O 18的振动状态的能量O与文献(可用于低振动激励)非常一致。此处获得的有关高激发振动态的新数据使我们能够对臭氧对称和非对称异构体中的科里奥利效应进行首次系统研究。









































 京公网安备 11010802027423号
京公网安备 11010802027423号