当前位置:
X-MOL 学术
›
Bioorgan. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Design, synthesis and anticancer activity of novel valproic acid conjugates with improved histone deacetylase (HDAC) inhibitory activity.
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2020-03-30 , DOI: 10.1016/j.bioorg.2020.103797 Tarek S Ibrahim 1 , Taghreed A Sheha 2 , Nader E Abo-Dya 3 , Mohammed A AlAwadh 4 , Nabil A Alhakamy 5 , Zakaria K Abdel-Samii 2 , Siva S Panda 6 , Gamal El-Din A Abuo-Rahma 7 , Mamdouh F A Mohamed 8
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2020-03-30 , DOI: 10.1016/j.bioorg.2020.103797 Tarek S Ibrahim 1 , Taghreed A Sheha 2 , Nader E Abo-Dya 3 , Mohammed A AlAwadh 4 , Nabil A Alhakamy 5 , Zakaria K Abdel-Samii 2 , Siva S Panda 6 , Gamal El-Din A Abuo-Rahma 7 , Mamdouh F A Mohamed 8
Affiliation
|
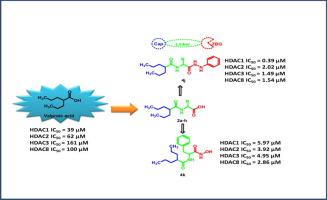
Twenty-five valproic acid conjugates have been designed and synthesized. All target compounds were explored for their in vitro anti-proliferative activities using the MTT-based assay against four human cancer cell lines includingliver (HePG2), colon (HCT116), breast (MCF7) and cervical (HeLa) carcinoma cell lines. Out of six valproic acid-amino acid conjugates 2a-f. Only cysteine containing conjugate 2f showed the significant activity (IC50 9.10 µM against HePG2 and 6.81 µM against HCT116). However conjugate 2j showed broad-spectrum antitumor activity against all cell lines tested. In addition, conjugates 4j and 4k which contains phenyl hydrazide and hydroxamic acid group, respectively, also showed broad spectrum activity. Furthermore, six compounds were screened for HDAC 1-9 isozymes inhibitory activities. Compounds 2j, 4j and 4k manifested a higher inhibitory activity more than valproic acid but less than SAHA. In addition, the in vivo antitumor screening of 2j, 4j and 4k was done and the results have shown that 2j, 4j and 4k, particularly 4j, showed a significant decrease in tumor size and presented a considerable decrease in viable EAC count. Docking study of selectedcompound 4j revealed that it can bind nicely to the binding pocket of HDAC 1, 2, 3, 4 and HDAC 8. The results suggest that compounds 2j, 4j and 4k, particularly 4j, may be promising lead candidates for the development of novel targeted anti-tumor drug potentially via inhibiting HDACs.
中文翻译:

具有改进的组蛋白脱乙酰基酶(HDAC)抑制活性的新型丙戊酸缀合物的设计,合成和抗癌活性。
已经设计和合成了二十五个丙戊酸缀合物。使用基于MTT的检测方法针对所有四种人类癌细胞系,包括肝(HePG2),结肠(HCT116),乳腺癌(MCF7)和宫颈(HeLa)癌细胞系,探索了所有目标化合物的体外抗增殖活性。在六个丙戊酸-氨基酸缀合物2a-f中。只有含半胱氨酸的结合物2f表现出显着的活性(针对HePG2的IC50为9.10 µM,针对HCT116的IC50为6.81 µM)。然而,缀合物2j对所有测试的细胞系显示广谱抗肿瘤活性。另外,分别包含苯酰肼和异羟肟酸基团的缀合物4j和4k也显示出广谱活性。此外,针对HDAC 1-9同工酶抑制活性筛选了6种化合物。化合物2j 4j和4k表现出比丙戊酸更高的抑制活性,但比SAHA更低。此外,对2j,4j和4k进行了体内抗肿瘤筛选,结果表明2j,4j和4k(尤其是4j)显示出肿瘤大小显着减小,并且可行的EAC计数显着降低。所选化合物4j的对接研究表明,它可以很好地与HDAC 1、2、3、4和HDAC 8的结合口袋结合。结果表明,化合物2j,4j和4k(尤其是4j)可能是开发的有希望的先导候选物新型靶向抗肿瘤药物可能通过抑制HDAC来实现。表现出肿瘤大小的显着减少,并且存活EAC计数显着下降。所选化合物4j的对接研究表明,它可以很好地与HDAC 1、2、3、4和HDAC 8的结合口袋结合。结果表明,化合物2j,4j和4k(尤其是4j)可能是开发的有希望的先导候选物新型靶向抗肿瘤药物可能通过抑制HDAC来实现。表现出肿瘤大小的显着减少,并且存活EAC计数显着减少。所选化合物4j的对接研究表明,它可以很好地与HDAC 1、2、3、4和HDAC 8的结合口袋结合。结果表明,化合物2j,4j和4k(尤其是4j)可能是开发的有希望的先导候选物新型靶向抗肿瘤药物可能通过抑制HDAC来实现。
更新日期:2020-04-20
中文翻译:
具有改进的组蛋白脱乙酰基酶(HDAC)抑制活性的新型丙戊酸缀合物的设计,合成和抗癌活性。
已经设计和合成了二十五个丙戊酸缀合物。使用基于MTT的检测方法针对所有四种人类癌细胞系,包括肝(HePG2),结肠(HCT116),乳腺癌(MCF7)和宫颈(HeLa)癌细胞系,探索了所有目标化合物的体外抗增殖活性。在六个丙戊酸-氨基酸缀合物2a-f中。只有含半胱氨酸的结合物2f表现出显着的活性(针对HePG2的IC50为9.10 µM,针对HCT116的IC50为6.81 µM)。然而,缀合物2j对所有测试的细胞系显示广谱抗肿瘤活性。另外,分别包含苯酰肼和异羟肟酸基团的缀合物4j和4k也显示出广谱活性。此外,针对HDAC 1-9同工酶抑制活性筛选了6种化合物。化合物2j 4j和4k表现出比丙戊酸更高的抑制活性,但比SAHA更低。此外,对2j,4j和4k进行了体内抗肿瘤筛选,结果表明2j,4j和4k(尤其是4j)显示出肿瘤大小显着减小,并且可行的EAC计数显着降低。所选化合物4j的对接研究表明,它可以很好地与HDAC 1、2、3、4和HDAC 8的结合口袋结合。结果表明,化合物2j,4j和4k(尤其是4j)可能是开发的有希望的先导候选物新型靶向抗肿瘤药物可能通过抑制HDAC来实现。表现出肿瘤大小的显着减少,并且存活EAC计数显着下降。所选化合物4j的对接研究表明,它可以很好地与HDAC 1、2、3、4和HDAC 8的结合口袋结合。结果表明,化合物2j,4j和4k(尤其是4j)可能是开发的有希望的先导候选物新型靶向抗肿瘤药物可能通过抑制HDAC来实现。表现出肿瘤大小的显着减少,并且存活EAC计数显着减少。所选化合物4j的对接研究表明,它可以很好地与HDAC 1、2、3、4和HDAC 8的结合口袋结合。结果表明,化合物2j,4j和4k(尤其是4j)可能是开发的有希望的先导候选物新型靶向抗肿瘤药物可能通过抑制HDAC来实现。










































 京公网安备 11010802027423号
京公网安备 11010802027423号