当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Natural transition orbitals for complex two-component excited state calculations
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-03-27 , DOI: 10.1002/jcc.26196 Joseph M Kasper 1 , Xiaosong Li 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-03-27 , DOI: 10.1002/jcc.26196 Joseph M Kasper 1 , Xiaosong Li 1
Affiliation
|
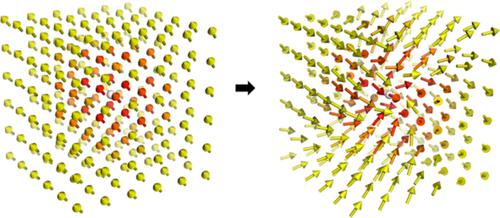
While the natural transition orbital (NTO) method has allowed electronic excitations from time‐dependent Hartree‐Fock and density functional theory to be viewed in a traditional orbital picture, the extension to multicomponent molecular orbitals such as those used in relativistic two‐component methods or generalized Hartree‐Fock (GHF) or generalized Kohn‐Sham (GKS) is less straightforward due to mixing of spin‐components and the inherent inclusion of spin‐flip transitions in time‐dependent GHF/GKS. An extension of single‐component NTOs to the two‐component framework is presented, in addition to a brief discussion of the practical aspects of visualizing two‐component complex orbitals. Unlike the single‐component analog, the method explicitly describes the spin and frequently obtains solutions with several significant orbital pairs. The method is presented using calculations on a mercury atom and a CrO2Cl2 complex.
中文翻译:

用于复杂双组分激发态计算的自然跃迁轨道
虽然自然跃迁轨道 (NTO) 方法允许在传统的轨道图片中查看来自时间相关 Hartree-Fock 和密度泛函理论的电子激发,但扩展到多组分分子轨道,例如在相对论双组分方法或广义 Hartree-Fock (GHF) 或广义 Kohn-Sham (GKS) 由于自旋分量的混合以及时间相关 GHF/GKS 中自旋翻转跃迁的固有包含而不太直接。除了对可视化双组件复杂轨道的实际方面的简要讨论之外,还介绍了单组件 NTO 到双组件框架的扩展。与单分量模拟不同,该方法明确描述了自旋,并经常获得具有几个重要轨道对的解。
更新日期:2020-03-27
中文翻译:
用于复杂双组分激发态计算的自然跃迁轨道
虽然自然跃迁轨道 (NTO) 方法允许在传统的轨道图片中查看来自时间相关 Hartree-Fock 和密度泛函理论的电子激发,但扩展到多组分分子轨道,例如在相对论双组分方法或广义 Hartree-Fock (GHF) 或广义 Kohn-Sham (GKS) 由于自旋分量的混合以及时间相关 GHF/GKS 中自旋翻转跃迁的固有包含而不太直接。除了对可视化双组件复杂轨道的实际方面的简要讨论之外,还介绍了单组件 NTO 到双组件框架的扩展。与单分量模拟不同,该方法明确描述了自旋,并经常获得具有几个重要轨道对的解。


























 京公网安备 11010802027423号
京公网安备 11010802027423号