当前位置:
X-MOL 学术
›
Int. J. Energy Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical research on p‐type doping two‐dimensional GaN based on first‐principles study
International Journal of Energy Research ( IF 4.3 ) Pub Date : 2020-03-27 , DOI: 10.1002/er.5380 Jian Tian 1 , Lei Liu 1 , Feifei Lu 1
International Journal of Energy Research ( IF 4.3 ) Pub Date : 2020-03-27 , DOI: 10.1002/er.5380 Jian Tian 1 , Lei Liu 1 , Feifei Lu 1
Affiliation
|
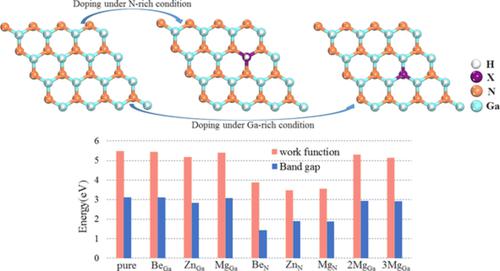
By using first principles, the p‐doping mechanism of two‐dimensional GaN with buckled structure is discussed in detail under various doping configurations, including different doping elements, positions, and concentrations. The research implies that difference in electronegativity between three doping elements: Be, Zn, Mg and two‐dimensional GaN results in a significant change in atomic structure and charge distribution. When Be, Zn, and Mg atoms are doped at Ga position, doping process in two‐dimensional GaN is easier because their formation energies are 1.684, 4.630, and 3.390 eV, respectively, which are lower than doped at N position. In addition, Ga doping site is more favorable for p‐type doping because bandgap and work function of two‐dimensional GaN are reduced and it would convert into p‐type semiconductor when a Ga atom is replaced by dopants.
中文翻译:

基于第一性原理的p型掺杂二维GaN的理论研究
通过使用第一原理,详细讨论了在各种掺杂配置下(包括不同的掺杂元素,位置和浓度)具有扭曲结构的二维GaN的p掺杂机理。研究表明,Be,Zn,Mg和二维GaN这三种掺杂元素之间的电负性差异会导致原子结构和电荷分布发生重大变化。当在Ga位置掺杂Be,Zn和Mg原子时,二维GaN中的掺杂过程更容易,因为它们的形成能分别为1.684、4.630和3.390 eV,这比在N位置掺杂的能量低。此外,
更新日期:2020-03-27
中文翻译:
基于第一性原理的p型掺杂二维GaN的理论研究
通过使用第一原理,详细讨论了在各种掺杂配置下(包括不同的掺杂元素,位置和浓度)具有扭曲结构的二维GaN的p掺杂机理。研究表明,Be,Zn,Mg和二维GaN这三种掺杂元素之间的电负性差异会导致原子结构和电荷分布发生重大变化。当在Ga位置掺杂Be,Zn和Mg原子时,二维GaN中的掺杂过程更容易,因为它们的形成能分别为1.684、4.630和3.390 eV,这比在N位置掺杂的能量低。此外,










































 京公网安备 11010802027423号
京公网安备 11010802027423号