Nature Communications ( IF 14.7 ) Pub Date : 2020-03-26 , DOI: 10.1038/s41467-020-15194-z Saori Sakaue 1, 2, 3 , Jun Hirata 1, 4 , Masahiro Kanai 1, 2, 5 , Ken Suzuki 1 , Masato Akiyama 2, 6 , Chun Lai Too 7, 8 , Thurayya Arayssi 9 , Mohammed Hammoudeh 10 , Samar Al Emadi 10 , Basel K Masri 11 , Hussein Halabi 12 , Humeira Badsha 13 , Imad W Uthman 14 , Richa Saxena 15, 16 , Leonid Padyukov 8 , Makoto Hirata 17 , Koichi Matsuda 18 , Yoshinori Murakami 19 , Yoichiro Kamatani 2, 20 , Yukinori Okada 1, 21, 22
|
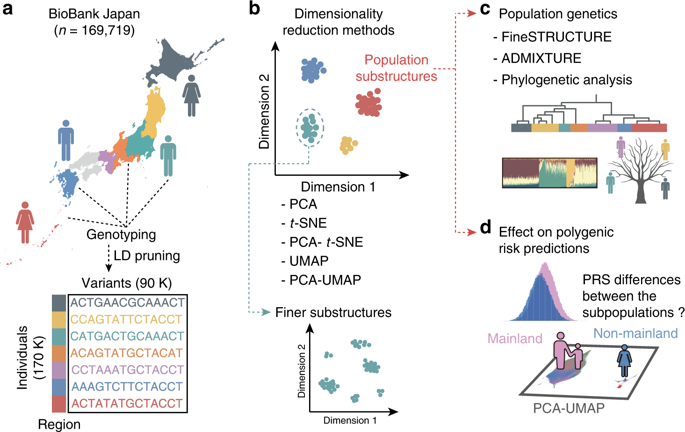
The diversity in our genome is crucial to understanding the demographic history of worldwide populations. However, we have yet to know whether subtle genetic differences within a population can be disentangled, or whether they have an impact on complex traits. Here we apply dimensionality reduction methods (PCA, t-SNE, PCA-t-SNE, UMAP, and PCA-UMAP) to biobank-derived genomic data of a Japanese population (n = 169,719). Dimensionality reduction reveals fine-scale population structure, conspicuously differentiating adjacent insular subpopulations. We further enluciate the demographic landscape of these Japanese subpopulations using population genetics analyses. Finally, we perform phenome-wide polygenic risk score (PRS) analyses on 67 complex traits. Differences in PRS between the deconvoluted subpopulations are not always concordant with those in the observed phenotypes, suggesting that the PRS differences might reflect biases from the uncorrected structure, in a trait-dependent manner. This study suggests that such an uncorrected structure can be a potential pitfall in the clinical application of PRS.
中文翻译:
降维揭示了日本人口的精细结构,对多基因风险预测产生影响。
我们基因组的多样性对于了解全球人口的人口历史至关重要。然而,我们还不知道群体内细微的遗传差异是否可以被消除,或者它们是否会对复杂的性状产生影响。在这里,我们将降维方法(PCA、 t -SNE、PCA -t -SNE、UMAP 和 PCA-UMAP)应用于来自生物库的日本人群基因组数据( n = 169,719)。降维揭示了精细尺度的种群结构,显着地区分了相邻的岛屿亚种群。我们利用群体遗传学分析进一步阐明这些日本亚人群的人口状况。最后,我们对 67 个复杂性状进行了全表型多基因风险评分 (PRS) 分析。去卷积亚群之间的 PRS 差异并不总是与观察到的表型中的差异一致,这表明 PRS 差异可能以性状依赖的方式反映了未校正结构的偏差。这项研究表明,这种未经纠正的结构可能是 PRS 临床应用中的潜在陷阱。










































 京公网安备 11010802027423号
京公网安备 11010802027423号