当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Gaussian Basis Sets for Crystalline Solids: All-Purpose Basis Set Libraries vs System-Specific Optimizations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-26 , DOI: 10.1021/acs.jctc.9b01004 Loredana Edith Daga 1 , Bartolomeo Civalleri 1 , Lorenzo Maschio 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-26 , DOI: 10.1021/acs.jctc.9b01004 Loredana Edith Daga 1 , Bartolomeo Civalleri 1 , Lorenzo Maschio 1
Affiliation
|
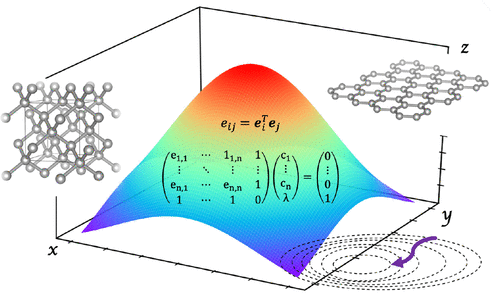
It is customary in molecular quantum chemistry to adopt basis set libraries in which the basis sets are classified according to either their size (triple-ζ, quadruple-ζ, ...) and the method/property they are optimal for (correlation-consistent, linear-response, ...) but not according to the chemistry of the system to be studied. In fact the vast majority of molecules is quite homogeneous in terms of density (i.e., atomic distances) and types of bond involved (covalent or dispersive). The situation is not the same for solids, in which the same chemical element can be found having metallic, ionic, covalent, or dispersively bound character in different crystalline forms or compounds, with different packings. This situation calls for a different approach to the choice of basis sets, namely a system-specific optimization of the basis set that requires a practical algorithm that could be used on a routine basis. In this work we develop a basis set optimization method based on an algorithm–similar to the direct inversion in the iterative subspace–that we name BDIIS. The total energy of the system is minimized together with the condition number of the overlap matrix as proposed by VandeVondele et al. [VandeVondele et al. J. Chem. Phys. 2007, 227, 114105]. The details of the method are here presented, and its performance in optimizing valence orbitals is shown. As demonstrative systems we consider simple prototypical solids such as diamond, graphene sodium chloride, and LiH, and we show how basis set optimizations have certain advantages also toward the use of large (quadruple-ζ) basis sets in solids, both at the DFT and Hartree–Fock level.
中文翻译:

结晶固体的高斯基集:通用基集库与系统特定的优化
在分子量子化学中通常采用基集库,其中基集根据它们的大小(三元-ζ,四元-ζ,...)和它们最适合的方法/属性(相关一致)进行分类。 ,线性响应,...),但不取决于要研究的系统的化学性质。实际上,就密度(即原子距离)和所涉及的键的类型(共价键或分散键)而言,绝大多数分子是相当均质的。对于固体而言,情况就不一样了,在其中可以发现相同的化学元素具有金属,离子,共价或分散键合特征,且具有不同的结晶形式或化合物,并具有不同的堆积。这种情况要求选择不同的基础集,即对基础集的特定于系统的优化,该优化要求可以在常规基础上使用的实用算法。在这项工作中,我们开发了一种基于算法的基础集优化方法,类似于迭代子空间中的直接反演,我们将其命名为BDIIS。如VandeVondele等人提出的,系统的总能量与重叠矩阵的条件数一起被最小化。[VandeVondele等。J.化学 物理 2007年, 227,114105]。这里介绍了该方法的详细信息,并显示了其在优化价轨道方面的性能。作为演示系统,我们考虑简单的原型固体,例如钻石,石墨烯氯化钠和LiH,并且我们展示了基集优化对于在DFT和DFT上在固体中使用大(四倍-ζ)基集也具有某些优势。 Hartree–Fock级。
更新日期:2020-04-24
中文翻译:
结晶固体的高斯基集:通用基集库与系统特定的优化
在分子量子化学中通常采用基集库,其中基集根据它们的大小(三元-ζ,四元-ζ,...)和它们最适合的方法/属性(相关一致)进行分类。 ,线性响应,...),但不取决于要研究的系统的化学性质。实际上,就密度(即原子距离)和所涉及的键的类型(共价键或分散键)而言,绝大多数分子是相当均质的。对于固体而言,情况就不一样了,在其中可以发现相同的化学元素具有金属,离子,共价或分散键合特征,且具有不同的结晶形式或化合物,并具有不同的堆积。这种情况要求选择不同的基础集,即对基础集的特定于系统的优化,该优化要求可以在常规基础上使用的实用算法。在这项工作中,我们开发了一种基于算法的基础集优化方法,类似于迭代子空间中的直接反演,我们将其命名为BDIIS。如VandeVondele等人提出的,系统的总能量与重叠矩阵的条件数一起被最小化。[VandeVondele等。J.化学 物理 2007年, 227,114105]。这里介绍了该方法的详细信息,并显示了其在优化价轨道方面的性能。作为演示系统,我们考虑简单的原型固体,例如钻石,石墨烯氯化钠和LiH,并且我们展示了基集优化对于在DFT和DFT上在固体中使用大(四倍-ζ)基集也具有某些优势。 Hartree–Fock级。









































 京公网安备 11010802027423号
京公网安备 11010802027423号