当前位置:
X-MOL 学术
›
Appl. Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
DFT study of n-alkyl carboxylic acids on oxidized aluminum surfaces: from standalone molecules to self-assembled-monolayers
Applied Surface Science ( IF 6.3 ) Pub Date : 2020-09-01 , DOI: 10.1016/j.apsusc.2020.146156 Matic Poberžnik , Fatah Chiter , Ingrid Milošev , Philippe Marcus , Dominique Costa , Anton Kokalj
Applied Surface Science ( IF 6.3 ) Pub Date : 2020-09-01 , DOI: 10.1016/j.apsusc.2020.146156 Matic Poberžnik , Fatah Chiter , Ingrid Milošev , Philippe Marcus , Dominique Costa , Anton Kokalj
|
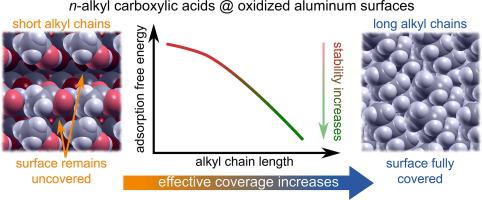
Abstract We report on a systematic density-functional-theory (DFT) study of adsorption of n-alkyl carboxylic acids (CA) on oxidized aluminum surfaces, where we address the roles of the adsorption mode, molecular coverage, tilt angle of alkyl chain, and alkyl chain size—from CA-2 to CA-18, where the suffix represents the number of C atoms in the molecule—on the stability of the formed CA monolayers. Adsorption was modeled on two models of the oxidized aluminum surface: both consist of a hydroxylated ultrathin-oxide film on top of Al(1 1 1). The two models differ in OH coverage, types of adsorption sites, and their lateral distribution. Two different adsorption modes were considered: (i) plain adsorption mode, where the CA binds to the surface with H-bonds, and (ii) acid-base condensation adsorption mode, where CA replaces a surface OH group by forming a water molecule as a co-product. Carboxylates bind considerably stronger to the surface than intact CAs, but due to bond-breaking energy cost, the adsorption free energy of the condensation mode is by about 0.15 eV less exergonic than that of the plain mode. While on the fully hydroxylated surface we only identified monodentate bonded carboxylates, bidentates are by about 0.7 eV more stable than monodentates near OH vacancies. Molecular packing within the monolayer is driven by lateral interchain interactions and the optimum tilt angle is achieved when the interchain distances are similar to those in the polyethylene crystal. This implies that the tilt angle depends on the coverage, i.e., it decreases with increasing coverage. At full monolayer coverage, the tilt angles are found to be about 40°–50° vs. the surface-normal, depending on the oxide/hydroxide composition on Al, and do not depend on chain size for longer chains (e.g., for CA-8 and beyond). The adsorption energy can be decomposed into the headgroup–surface and lateral interchain components, the former being largely independent on the chain size and the latter being proportional to the number of C atoms in the chain. Consequently, the adsorption is stabilized by about 1 eV/molecule at full monolayer coverage as passing from CA-2 to CA-18. Finally, we propose a simplified way to calculate the vibrational contribution to the free energy of the adsorbed monolayers.
中文翻译:

氧化铝表面正烷基羧酸的 DFT 研究:从独立分子到自组装单层
摘要 我们报告了正烷基羧酸 (CA) 在氧化铝表面吸附的系统密度泛函理论 (DFT) 研究,其中我们解决了吸附模式、分子覆盖率、烷基链的倾斜角、和烷基链的大小——从 CA-2 到 CA-18,其中后缀代表分子中 C 原子的数量——对形成的 CA 单层的稳定性。吸附模拟了氧化铝表面的两种模型:均由 Al(1 1 1) 顶部的羟基化超薄氧化膜组成。这两种模型在 OH 覆盖率、吸附位点类型及其横向分布方面有所不同。考虑了两种不同的吸附模式:(i)普通吸附模式,其中 CA 通过 H 键结合到表面,以及(ii)酸碱缩合吸附模式,其中 CA 通过形成水分子作为副产物来取代表面 OH 基团。羧酸盐与表面的结合比完整的 CA 强得多,但由于键断裂能量成本,缩合模式的吸附自由能比普通模式低约 0.15 eV。虽然在完全羟基化的表面上,我们只发现了单齿键合的羧酸盐,但双齿比靠近 OH 空位的单齿稳定约 0.7 eV。单层内的分子堆积由横向链间相互作用驱动,当链间距离与聚乙烯晶体中的距离相似时,可实现最佳倾斜角。这意味着倾斜角取决于覆盖范围,即它随着覆盖范围的增加而减小。在完全单层覆盖时,发现倾斜角与表面法线成大约 40°–50°,这取决于 Al 上的氧化物/氢氧化物成分,并且不取决于更长链的链尺寸(例如,对于 CA-8 及以上) . 吸附能可以分解为头基-表面和横向链间成分,前者在很大程度上与链大小无关,后者与链中 C 原子的数量成正比。因此,当从 CA-2 传递到 CA-18 时,在完全单层覆盖下,吸附稳定了约 1 eV/分子。最后,我们提出了一种计算吸附单层自由能的振动贡献的简化方法。吸附能可以分解为头基-表面和横向链间成分,前者在很大程度上与链大小无关,后者与链中 C 原子的数量成正比。因此,当从 CA-2 传递到 CA-18 时,在完全单层覆盖下,吸附稳定了约 1 eV/分子。最后,我们提出了一种简化的方法来计算对吸附单层自由能的振动贡献。吸附能可以分解为头基-表面和横向链间成分,前者在很大程度上与链大小无关,后者与链中 C 原子的数量成正比。因此,当从 CA-2 传递到 CA-18 时,在完全单层覆盖下,吸附稳定了约 1 eV/分子。最后,我们提出了一种计算吸附单层自由能的振动贡献的简化方法。
更新日期:2020-09-01
中文翻译:
氧化铝表面正烷基羧酸的 DFT 研究:从独立分子到自组装单层
摘要 我们报告了正烷基羧酸 (CA) 在氧化铝表面吸附的系统密度泛函理论 (DFT) 研究,其中我们解决了吸附模式、分子覆盖率、烷基链的倾斜角、和烷基链的大小——从 CA-2 到 CA-18,其中后缀代表分子中 C 原子的数量——对形成的 CA 单层的稳定性。吸附模拟了氧化铝表面的两种模型:均由 Al(1 1 1) 顶部的羟基化超薄氧化膜组成。这两种模型在 OH 覆盖率、吸附位点类型及其横向分布方面有所不同。考虑了两种不同的吸附模式:(i)普通吸附模式,其中 CA 通过 H 键结合到表面,以及(ii)酸碱缩合吸附模式,其中 CA 通过形成水分子作为副产物来取代表面 OH 基团。羧酸盐与表面的结合比完整的 CA 强得多,但由于键断裂能量成本,缩合模式的吸附自由能比普通模式低约 0.15 eV。虽然在完全羟基化的表面上,我们只发现了单齿键合的羧酸盐,但双齿比靠近 OH 空位的单齿稳定约 0.7 eV。单层内的分子堆积由横向链间相互作用驱动,当链间距离与聚乙烯晶体中的距离相似时,可实现最佳倾斜角。这意味着倾斜角取决于覆盖范围,即它随着覆盖范围的增加而减小。在完全单层覆盖时,发现倾斜角与表面法线成大约 40°–50°,这取决于 Al 上的氧化物/氢氧化物成分,并且不取决于更长链的链尺寸(例如,对于 CA-8 及以上) . 吸附能可以分解为头基-表面和横向链间成分,前者在很大程度上与链大小无关,后者与链中 C 原子的数量成正比。因此,当从 CA-2 传递到 CA-18 时,在完全单层覆盖下,吸附稳定了约 1 eV/分子。最后,我们提出了一种计算吸附单层自由能的振动贡献的简化方法。吸附能可以分解为头基-表面和横向链间成分,前者在很大程度上与链大小无关,后者与链中 C 原子的数量成正比。因此,当从 CA-2 传递到 CA-18 时,在完全单层覆盖下,吸附稳定了约 1 eV/分子。最后,我们提出了一种简化的方法来计算对吸附单层自由能的振动贡献。吸附能可以分解为头基-表面和横向链间成分,前者在很大程度上与链大小无关,后者与链中 C 原子的数量成正比。因此,当从 CA-2 传递到 CA-18 时,在完全单层覆盖下,吸附稳定了约 1 eV/分子。最后,我们提出了一种计算吸附单层自由能的振动贡献的简化方法。










































 京公网安备 11010802027423号
京公网安备 11010802027423号