当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Flexible Fitting of Small Molecules into Electron Microscopy Maps Using Molecular Dynamics Simulations with Neural Network Potentials.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-24 , DOI: 10.1021/acs.jcim.9b01167 John W Vant 1 , Shae-Lynn J Lahey 2 , Kalyanashis Jana 3 , Mrinal Shekhar 1, 4 , Daipayan Sarkar 1 , Barbara H Munk 1 , Ulrich Kleinekathöfer 3 , Sumit Mittal 1, 5 , Christopher Rowley 2 , Abhishek Singharoy 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-03-24 , DOI: 10.1021/acs.jcim.9b01167 John W Vant 1 , Shae-Lynn J Lahey 2 , Kalyanashis Jana 3 , Mrinal Shekhar 1, 4 , Daipayan Sarkar 1 , Barbara H Munk 1 , Ulrich Kleinekathöfer 3 , Sumit Mittal 1, 5 , Christopher Rowley 2 , Abhishek Singharoy 1
Affiliation
|
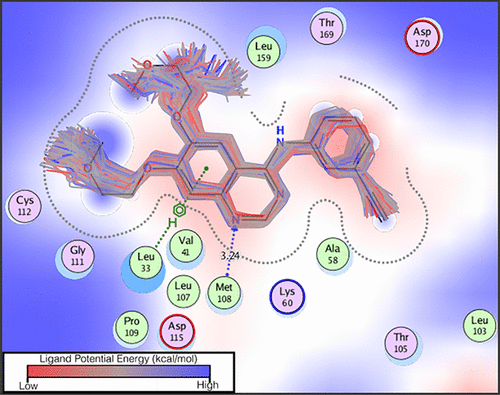
Despite significant advances in resolution, the potential for cryo-electron microscopy (EM) to be used in determining the structures of protein-drug complexes remains unrealized. Determination of accurate structures and coordination of bound ligands necessitates simultaneous fitting of the models into the density envelopes, exhaustive sampling of the ligand geometries, and, most importantly, concomitant rearrangements in the side chains to optimize the binding energy changes. In this article, we present a flexible-fitting pipeline where molecular dynamics flexible fitting (MDFF) is used to refine structures of protein-ligand complexes from 3 to 5 Å electron density data. Enhanced sampling is employed to explore the binding pocket rearrangements. To provide a model that can accurately describe the conformational dynamics of the chemically diverse set of small-molecule drugs inside MDFF, we use QM/MM and neural-network potential (NNP)/MM models of protein-ligand complexes, where the ligand is represented using the QM or NNP model, and the protein is represented using established molecular mechanical force fields (e.g., CHARMM). This pipeline offers structures commensurate to or better than recently submitted high-resolution cryo-EM or X-ray models, even when given medium to low-resolution data as input. The use of the NNPs makes the algorithm more robust to the choice of search models, offering a radius of convergence of 6.5 Å for ligand structure determination. The quality of the predicted structures was also judged by density functional theory calculations of ligand strain energy. This strain potential energy is found to systematically decrease with better fitting to density and improved ligand coordination, indicating correct binding interactions. A computationally inexpensive protocol for computing strain energy is reported as part of the model analysis protocol that monitors both the ligand fit as well as model quality.
中文翻译:

使用具有神经网络势的分子动力学模拟将小分子灵活地拟合到电子显微镜图中。
尽管分辨率取得了显着进步,但冷冻电子显微镜(EM)用于确定蛋白质-药物复合物结构的潜力仍未实现。确定结合配体的精确结构和配位需要同时将模型拟合到密度包络中,对配体几何形状进行详尽采样,最重要的是,侧链中的伴随重排以优化结合能变化。在本文中,我们提出了一种柔性拟合流程,其中分子动力学柔性拟合 (MDFF) 用于根据 3 至 5 Å 电子密度数据精炼蛋白质-配体复合物的结构。采用增强采样来探索结合口袋重排。为了提供一个能够准确描述 MDFF 内化学多样化的小分子药物构象动力学的模型,我们使用蛋白质-配体复合物的 QM/MM 和神经网络电位 (NNP)/MM 模型,其中配体是使用QM或NNP模型表示,并且使用已建立的分子机械力场(例如CHARMM)表示蛋白质。该管道提供的结构与最近提交的高分辨率冷冻电镜或 X 射线模型相当或更好,即使以中低分辨率数据作为输入也是如此。 NNP 的使用使算法对搜索模型的选择更加鲁棒,为配体结构确定提供了 6.5 Å 的收敛半径。预测结构的质量还通过配体应变能的密度泛函理论计算来判断。发现这种应变势能随着密度的更好拟合和配体协调的改善而系统地降低,表明正确的结合相互作用。 据报道,用于计算应变能的计算成本低廉的协议作为模型分析协议的一部分,用于监测配体拟合和模型质量。
更新日期:2020-03-24
中文翻译:
使用具有神经网络势的分子动力学模拟将小分子灵活地拟合到电子显微镜图中。
尽管分辨率取得了显着进步,但冷冻电子显微镜(EM)用于确定蛋白质-药物复合物结构的潜力仍未实现。确定结合配体的精确结构和配位需要同时将模型拟合到密度包络中,对配体几何形状进行详尽采样,最重要的是,侧链中的伴随重排以优化结合能变化。在本文中,我们提出了一种柔性拟合流程,其中分子动力学柔性拟合 (MDFF) 用于根据 3 至 5 Å 电子密度数据精炼蛋白质-配体复合物的结构。采用增强采样来探索结合口袋重排。为了提供一个能够准确描述 MDFF 内化学多样化的小分子药物构象动力学的模型,我们使用蛋白质-配体复合物的 QM/MM 和神经网络电位 (NNP)/MM 模型,其中配体是使用QM或NNP模型表示,并且使用已建立的分子机械力场(例如CHARMM)表示蛋白质。该管道提供的结构与最近提交的高分辨率冷冻电镜或 X 射线模型相当或更好,即使以中低分辨率数据作为输入也是如此。 NNP 的使用使算法对搜索模型的选择更加鲁棒,为配体结构确定提供了 6.5 Å 的收敛半径。预测结构的质量还通过配体应变能的密度泛函理论计算来判断。发现这种应变势能随着密度的更好拟合和配体协调的改善而系统地降低,表明正确的结合相互作用。 据报道,用于计算应变能的计算成本低廉的协议作为模型分析协议的一部分,用于监测配体拟合和模型质量。










































 京公网安备 11010802027423号
京公网安备 11010802027423号