JAMA Neurology ( IF 20.4 ) Pub Date : 2020-06-01 , DOI: 10.1001/jamaneurol.2020.0299 Blair R Leavitt 1 , Holly B Kordasiewicz 2 , Scott A Schobel 3
|
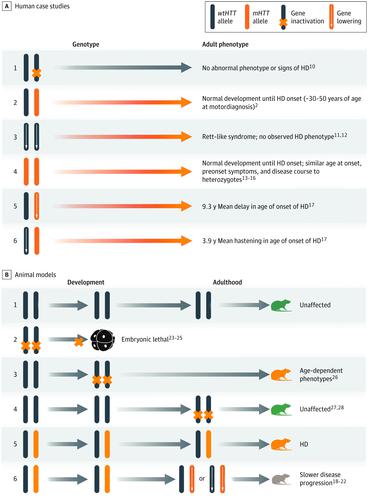
Huntington disease (HD) is caused by a cytosine-adenine-guanine trinucleotide repeat expansion in the huntingtin gene, HTT, that results in expression of variant (mutant) huntingtin protein (HTT). Therapeutic strategies that reduce HTT levels are currently being pursued to slow or stop disease progression in people with HD. These approaches are supported by robust preclinical data indicating that reducing variant huntingtin protein is associated with decreased HD pathology. However, the risk-benefit profile of reducing either variant HTT or both variant and wild-type HTT is currently an open question that is being addressed in ongoing clinical trials. This review aims to examine the current data available regarding altered HTT in humans, normal animals, and animal models of HD. Studies indexed in PubMed were searched using the MeSH term Huntington disease or the text words huntington or huntingtin from August 31, 1999, to August 31, 2019, with no language restrictions. Additional studies were included from the reference lists of relevant studies and the authors’ personal files. Articles describing at least 1 aspect of HTT reduction were included, prioritizing those published within the last 10 years. In vivo studies were also prioritized, with a focus on studies that examined the consequences of wild-type HTT reduction in adults. In a recently completed phase 1/2a study of RG6042 in 46 adults with early manifest HD, antisense oligonucleotide-mediated partial reduction of HTT was reported to be generally safe and well tolerated over the course of 4-monthly RG6042 doses. In case studies of people with rare genetic variations in huntingtin alleles, the loss of 1 wild-type allele was not associated with HD. People with homozygous cytosine-adenine-guanine expansions developed normally until the onset of HD, although they may have experienced a more aggressive disease course. In mouse models of HD, partial reduction of HTT was beneficial, with improvements in motor, cognitive, and behavioral phenotypes. The partial reduction of wild-type HTT in normal adult rodents and nonhuman primates was generally safe and well tolerated. The body of evidence reviewed in this article indicates a positive risk-benefit profile for the partial reduction of either variant HTT alone or both variant and wild-type HTT. These strategies target the underlying cause of HD and are currently being tested in several investigational clinical trials.
中文翻译:
降低亨廷顿病的亨廷顿疗法:潜在收益和风险的证据综述。
亨廷顿病(HD)是由亨廷顿基因HTT中的胞嘧啶-腺嘌呤-鸟嘌呤三核苷酸重复序列扩增引起的,该突变导致亨廷顿蛋白(HTT)变体表达。目前正在寻求降低HTT水平的治疗策略,以减缓或阻止HD人群的疾病进展。这些方法得到了可靠的临床前数据的支持,这些数据表明减少变体亨廷顿蛋白与降低的HD病理学有关。但是,降低变体HTT或同时降低变体HTT和野生型HTT的风险-收益状况目前是一个悬而未决的问题,正在进行的临床试验中正在解决。这项审查旨在检查有关改变HTT的当前可用数据在人类,正常动物和高清动物模型中。使用MeSH术语“亨廷顿病”或文字“亨廷顿或亨廷顿”来搜索PubMed中索引的研究从1999年8月31日到2019年8月31日,没有语言限制。其他研究也包括相关研究的参考清单和作者的个人档案。包括描述HTT降低至少一个方面的文章,优先考虑最近10年内发表的文章。还优先进行了体内研究,重点是研究了成人野生型HTT降低的后果的研究。在最近完成的对早期表现为HD的46位成年人的RG6042的1 / 2a期研究中,据报道,在每4个月RG6042剂量的过程中,反义寡核苷酸介导的HTT的部分降低通常是安全的,并且耐受性良好。在亨廷顿等位基因中遗传变异少的人的案例研究中,丢失1个野生型等位基因与HD无关。具有纯合的胞嘧啶-腺嘌呤-鸟嘌呤扩增的人正常发育直至HD发作,尽管他们可能经历了更具侵略性的疾病过程。在HD小鼠模型中,HTT的部分降低是有益的,可以改善运动,认知和行为表型。在正常的成年啮齿动物和非人类灵长类动物中,野生型HTT的部分降低通常是安全的,并且具有良好的耐受性。本文所审查的证据表明,单独降低变体HTT或降低变体HTT和野生型HTT的风险收益均为正。这些策略针对HD的根本原因,目前正在一些临床研究试验中进行测试。在HD小鼠模型中,HTT的部分降低是有益的,可以改善运动,认知和行为表型。在正常的成年啮齿动物和非人类灵长类动物中,野生型HTT的部分降低通常是安全的,并且具有良好的耐受性。本文审查的证据表明,单独降低变体HTT或降低变体HTT和野生型HTT均能部分降低风险的正面收益。这些策略针对HD的根本原因,目前正在一些临床研究试验中进行测试。在HD小鼠模型中,HTT的部分降低是有益的,可以改善运动,认知和行为表型。在正常的成年啮齿动物和非人类灵长类动物中,野生型HTT的部分降低通常是安全的,并且具有良好的耐受性。本文审查的证据表明,单独降低变体HTT或降低变体HTT和野生型HTT均能部分降低风险的正面收益。这些策略针对HD的根本原因,目前正在一些临床研究试验中进行测试。本文审查的证据表明,单独降低变体HTT或降低变体HTT和野生型HTT均能部分降低风险的正面收益。这些策略针对HD的根本原因,目前正在一些临床研究试验中进行测试。本文审查的证据表明,单独降低变体HTT或降低变体HTT和野生型HTT均能部分降低风险的正面收益。这些策略针对HD的根本原因,目前正在一些临床研究试验中进行测试。










































 京公网安备 11010802027423号
京公网安备 11010802027423号