当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Mechanistic Insights into the Origin of Stereoselectivity in an Asymmetric Chlorolactonization Catalyzed by (DHQD)2PHAL
Journal of the American Chemical Society ( IF 15.0 ) Pub Date : 2020-03-21 , DOI: 10.1021/jacs.0c01830 Roozbeh Yousefi 1 , Aritra Sarkar 1 , Kumar Dilip Ashtekar 1 , Daniel C. Whitehead 1 , Tayeb Kakeshpour 1 , Daniel Holmes 1 , Paul Reed 1 , James E. Jackson 1 , Babak Borhan 1
Journal of the American Chemical Society ( IF 15.0 ) Pub Date : 2020-03-21 , DOI: 10.1021/jacs.0c01830 Roozbeh Yousefi 1 , Aritra Sarkar 1 , Kumar Dilip Ashtekar 1 , Daniel C. Whitehead 1 , Tayeb Kakeshpour 1 , Daniel Holmes 1 , Paul Reed 1 , James E. Jackson 1 , Babak Borhan 1
Affiliation
|
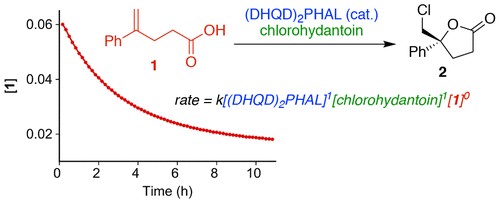
Electrophilic halofunctionalization reactions have undergone a resurgence sparked by recent discoveries in the field of catalytic asymmet-ric halocyclizations. To build mechanistic understanding of these asymmetric transformations, a toolbox of analytical methods has been deployed, addressing the roles of catalyst, electrophile (halenium donor), and nucleophile in determining rates and stereopreferences. The test reaction, (DHQD)2PHAL-catalyzed chlorocyclization of 4-aryl-4-pentenoic acids with 1,3-dichloro-5,5-dimethylhydantoin (DCDMH), is revealed to be first order in catalyst and chlorenium ion donor and zero order in alkenoic acid substrate under synthetically relevant conditions. The simplest interpretation is that rapid substrate-catalyst binding precedes rate-limiting chlorenium attack, controlling the face selectivity of both chlorine attack and lactone closure. ROESY and DFT studies, aided by crystal structures of carboxylic acids bound by the catalyst, point to a plausible resting state of the catalyst-substrate complex predisposed for asymmetric chlorolactoni-zation,. As revealed by our earlier labeling studies, these findings suggest modes of binding in the (DHQD)2PHAL chiral pocket that explain the system's remarkable control over rate- and enantioselection-determining events. Though a comprehensive modeling analysis is beyond the scope of the present work, quantum chemical analysis of the fragments' interactions and candidate reaction paths point to a one-step concerted process with the nucleophile playing a critical role in activating the olefin for concomitant electrophilic attack.
中文翻译:

对 (DHQD)2PHAL 催化的不对称氯内酯化中立体选择性起源的机理见解
最近在催化不对称卤环化领域的发现引发了亲电卤代官能化反应的复兴。为了建立对这些不对称转换的机械理解,已经部署了一个分析方法工具箱,解决了催化剂、亲电子试剂(卤供体)和亲核试剂在确定速率和立体偏好中的作用。测试反应,(DHQD)2PHAL 催化的 4-芳基-4-戊烯酸与 1,3-二氯-5,5-二甲基乙内酰脲 (DDCDMH) 的氯环化反应是催化剂和氯离子供体的一级反应,而零在合成相关条件下,烯酸底物的顺序。最简单的解释是快速底物-催化剂结合先于限速氯攻击,控制氯攻击和内酯关闭的表面选择性。ROESY 和 DFT 研究,借助与催化剂结合的羧酸的晶体结构,指出了倾向于不对称氯内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。ROESY 和 DFT 研究,借助与催化剂结合的羧酸的晶体结构,指出了倾向于不对称氯内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。ROESY 和 DFT 研究,借助与催化剂结合的羧酸的晶体结构,指出了倾向于不对称氯内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。指向倾向于不对称氯代内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析指出了一个一步协调的过程,亲核试剂在激活烯烃以进行伴随的亲电攻击方面起着关键作用。指向倾向于不对称氯代内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析指出了一个一步协调的过程,亲核试剂在激活烯烃以进行伴随的亲电攻击方面起着关键作用。这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了当前工作的范围,但片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了当前工作的范围,但片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。
更新日期:2020-03-21
中文翻译:
对 (DHQD)2PHAL 催化的不对称氯内酯化中立体选择性起源的机理见解
最近在催化不对称卤环化领域的发现引发了亲电卤代官能化反应的复兴。为了建立对这些不对称转换的机械理解,已经部署了一个分析方法工具箱,解决了催化剂、亲电子试剂(卤供体)和亲核试剂在确定速率和立体偏好中的作用。测试反应,(DHQD)2PHAL 催化的 4-芳基-4-戊烯酸与 1,3-二氯-5,5-二甲基乙内酰脲 (DDCDMH) 的氯环化反应是催化剂和氯离子供体的一级反应,而零在合成相关条件下,烯酸底物的顺序。最简单的解释是快速底物-催化剂结合先于限速氯攻击,控制氯攻击和内酯关闭的表面选择性。ROESY 和 DFT 研究,借助与催化剂结合的羧酸的晶体结构,指出了倾向于不对称氯内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。ROESY 和 DFT 研究,借助与催化剂结合的羧酸的晶体结构,指出了倾向于不对称氯内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。ROESY 和 DFT 研究,借助与催化剂结合的羧酸的晶体结构,指出了倾向于不对称氯内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。指向倾向于不对称氯代内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析指出了一个一步协调的过程,亲核试剂在激活烯烃以进行伴随的亲电攻击方面起着关键作用。指向倾向于不对称氯代内酯化的催化剂 - 底物复合物的合理静止状态。正如我们早期的标记研究所揭示的那样,这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了目前的工作范围,但对片段相互作用和候选反应路径的量子化学分析指出了一个一步协调的过程,亲核试剂在激活烯烃以进行伴随的亲电攻击方面起着关键作用。这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了当前工作的范围,但片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。这些发现表明 (DHQD)2PHAL 手性口袋中的结合模式解释了系统对速率和对映选择决定事件的显着控制。尽管全面的建模分析超出了当前工作的范围,但片段相互作用和候选反应路径的量子化学分析表明,亲核试剂在激活烯烃以进行伴随亲电攻击方面起着关键作用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号