当前位置:
X-MOL 学术
›
ACS Chem. Neurosci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Prediction of the Binding Affinities and Selectivity for CB1 and CB2 Ligands Using Homology Modeling, Molecular Docking, Molecular Dynamics Simulations, and MM-PBSA Binding Free Energy Calculations.
ACS Chemical Neuroscience ( IF 4.1 ) Pub Date : 2020-03-20 , DOI: 10.1021/acschemneuro.9b00696 Beihong Ji 1 , Shuhan Liu 1 , Xibing He 1 , Viet Hoang Man 1 , Xiang-Qun Xie 1 , Junmei Wang 1
ACS Chemical Neuroscience ( IF 4.1 ) Pub Date : 2020-03-20 , DOI: 10.1021/acschemneuro.9b00696 Beihong Ji 1 , Shuhan Liu 1 , Xibing He 1 , Viet Hoang Man 1 , Xiang-Qun Xie 1 , Junmei Wang 1
Affiliation
|
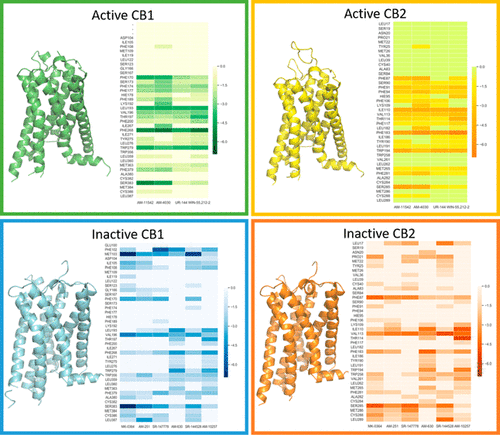
Cannabinoids are a group of chemical compounds that have been used for thousands of years due to their psychoactive function and systemic physiological effects. There are at least two types of cannabinoid receptors, CB1 and CB2, which belong to the G protein-coupled receptor superfamily and can trigger different signaling pathways to exert their physiological functions. In this study, several representative agonists and antagonists of both CB1 and CB2 were systematically studied to predict their binding affinities and selectivity against both cannabinoid receptors using a set of hierarchical molecular modeling and simulation techniques, including homology modeling, molecular docking, molecular dynamics (MD) simulations and end point binding free energy calculations using the molecular mechanics/Poisson–Boltzmann surface area-WSAS (MM-PBSA-WSAS) method, and molecular mechanics/generalized Born surface area (MM-GBSA) free energy decomposition. Encouragingly, the calculated binding free energies correlated very well with the experimental values and the correlation coefficient square (R2), 0.60, was much higher than that of an efficient but less accurate docking scoring function (R2 = 0.37). The hotspot residues for CB1 and CB2 in both active and inactive conformations were identified via MM-GBSA free energy decomposition analysis. The comparisons of binding free energies, ligand–receptor interaction patterns, and hotspot residues among the four systems, namely, agonist-bound CB1, agonist-bound CB2, antagonist-bound CB1, and antagonist-bound CB2, enabled us to investigate and identify distinct binding features of these four systems, with which one can rationally design potent, selective, and function-specific modulators for the cannabinoid receptors.
中文翻译:

使用同源建模、分子对接、分子动力学模拟和 MM-PBSA 结合自由能计算预测 CB1 和 CB2 配体的结合亲和力和选择性。
大麻素是一组化合物,由于其精神活性功能和全身生理效应,已经使用了数千年。至少有两种类型的大麻素受体,CB1和CB2,属于G蛋白偶联受体超家族,可以触发不同的信号通路发挥其生理功能。在这项研究中,系统地研究了 CB1 和 CB2 的几种代表性激动剂和拮抗剂,以使用一组分层分子建模和模拟技术(包括同源建模、分子对接、使用分子力学/泊松-玻尔兹曼表面积-WSAS (MM-PBSA-WSAS) 方法和分子力学/广义玻恩表面积 (MM-GBSA) 自由能分解的分子动力学 (MD) 模拟和端点结合自由能计算. 令人鼓舞的是,计算出的结合自由能与实验值和相关系数平方(R 2 ), 0.60,远高于高效但不太准确的对接评分函数 ( R 2 = 0.37)。通过MM-GBSA 自由能分解分析,确定了活性和非活性构象中 CB1 和 CB2 的热点残基。四种系统(即激动剂结合的 CB1、激动剂结合的 CB2、拮抗剂结合的 CB1 和拮抗剂结合的 CB2)的结合自由能、配体-受体相互作用模式和热点残基的比较,使我们能够研究和鉴定这四种系统的独特结合特征,可以利用这些特征合理地设计大麻素受体的有效、选择性和功能特异性调节剂。
更新日期:2020-04-23
中文翻译:
使用同源建模、分子对接、分子动力学模拟和 MM-PBSA 结合自由能计算预测 CB1 和 CB2 配体的结合亲和力和选择性。
大麻素是一组化合物,由于其精神活性功能和全身生理效应,已经使用了数千年。至少有两种类型的大麻素受体,CB1和CB2,属于G蛋白偶联受体超家族,可以触发不同的信号通路发挥其生理功能。在这项研究中,系统地研究了 CB1 和 CB2 的几种代表性激动剂和拮抗剂,以使用一组分层分子建模和模拟技术(包括同源建模、分子对接、使用分子力学/泊松-玻尔兹曼表面积-WSAS (MM-PBSA-WSAS) 方法和分子力学/广义玻恩表面积 (MM-GBSA) 自由能分解的分子动力学 (MD) 模拟和端点结合自由能计算. 令人鼓舞的是,计算出的结合自由能与实验值和相关系数平方(R 2 ), 0.60,远高于高效但不太准确的对接评分函数 ( R 2 = 0.37)。通过MM-GBSA 自由能分解分析,确定了活性和非活性构象中 CB1 和 CB2 的热点残基。四种系统(即激动剂结合的 CB1、激动剂结合的 CB2、拮抗剂结合的 CB1 和拮抗剂结合的 CB2)的结合自由能、配体-受体相互作用模式和热点残基的比较,使我们能够研究和鉴定这四种系统的独特结合特征,可以利用这些特征合理地设计大麻素受体的有效、选择性和功能特异性调节剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号