当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Detailed Pair Natural Orbital-Based Coupled Cluster Studies of Spin Crossover Energetics.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-20 , DOI: 10.1021/acs.jctc.9b01109 Benedikt M Flöser 1 , Yang Guo 2 , Christoph Riplinger 3 , Felix Tuczek 1 , Frank Neese 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-20 , DOI: 10.1021/acs.jctc.9b01109 Benedikt M Flöser 1 , Yang Guo 2 , Christoph Riplinger 3 , Felix Tuczek 1 , Frank Neese 2
Affiliation
|
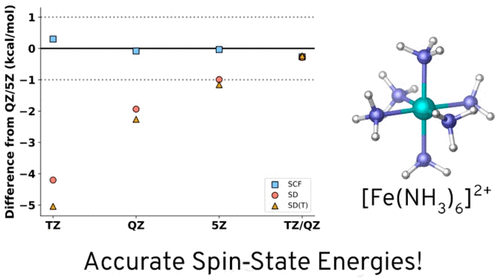
In this work, a detailed study of spin-state splittings in three spin crossover model compounds with DLPNO-CCSD(T) is presented. The performance in comparison to canonical CCSD(T) is assessed in detail. It was found that spin-state splittings with chemical accuracy, compared to the canonical results, are achieved when the full iterative triples (T1) scheme and TightPNO settings are applied and relativistic effects are taken into account. Having established the level of accuracy that can be reached relative to the canonical results, we have undertaken a detailed basis set study in the second part of the study. The slow convergence of the results of correlated calculations with respect to basis set extension is particularly acute for spin-state splittings for reasons discussed in detail in this Article. In fact, for some of the studied systems, 5Z basis sets are necessary in order to come close to the basis set limit that is estimated here by basis set extrapolation. Finally, the results of the present work are compared to available literature. In general, acceptable agreement with previous CCSD(T) results is found, although notable deviations stemming from differences in methodology and basis sets are noted. It is noted that the published CASPT2 numbers are far away from the extrapolated CCSD(T) numbers. In addition, dynamic quantum Monte Carlo results differ by several tens of kcal/mol from the CCSD(T) numbers. A comparison to DFT results produced with a range of popular density functionals shows the expected scattering of results and showcases the difficulty of applying DFT to spin-state energies.
中文翻译:

自旋交叉能量学的详细的基于自然轨道的耦合簇研究。
在这项工作中,采用 DLPNO-CCSD(T) 详细研究了三种自旋交叉模型化合物的自旋态分裂。详细评估了与规范 CCSD(T) 相比的性能。研究发现,与典型结果相比,当应用完整迭代三元组 (T 1 ) 方案和 TightPNO 设置并考虑相对论效应时,可以实现具有化学精度的自旋态分裂。在确定了相对于规范结果可以达到的准确性水平后,我们在研究的第二部分中进行了详细的基础集研究。由于本文详细讨论的原因,关于基组扩展的相关计算结果的缓慢收敛对于自旋态分裂尤其严重。事实上,对于一些研究的系统,5Z 基组是必要的,以便接近通过基组外推法估计的基组极限。最后,将本工作的结果与现有文献进行比较。总的来说,与之前的 CCSD(T) 结果存在可接受的一致性,尽管注意到由于方法和基础集的差异而产生的显着偏差。值得注意的是,公布的 CASPT2 数字与推断的 CCSD(T) 数字相差甚远。此外,动态量子蒙特卡罗结果与 CCSD(T) 数值相差数十 kcal/mol。与一系列流行的密度泛函产生的 DFT 结果的比较显示了预期的结果散射,并展示了将 DFT 应用到自旋态能量的难度。
更新日期:2020-04-24
中文翻译:
自旋交叉能量学的详细的基于自然轨道的耦合簇研究。
在这项工作中,采用 DLPNO-CCSD(T) 详细研究了三种自旋交叉模型化合物的自旋态分裂。详细评估了与规范 CCSD(T) 相比的性能。研究发现,与典型结果相比,当应用完整迭代三元组 (T 1 ) 方案和 TightPNO 设置并考虑相对论效应时,可以实现具有化学精度的自旋态分裂。在确定了相对于规范结果可以达到的准确性水平后,我们在研究的第二部分中进行了详细的基础集研究。由于本文详细讨论的原因,关于基组扩展的相关计算结果的缓慢收敛对于自旋态分裂尤其严重。事实上,对于一些研究的系统,5Z 基组是必要的,以便接近通过基组外推法估计的基组极限。最后,将本工作的结果与现有文献进行比较。总的来说,与之前的 CCSD(T) 结果存在可接受的一致性,尽管注意到由于方法和基础集的差异而产生的显着偏差。值得注意的是,公布的 CASPT2 数字与推断的 CCSD(T) 数字相差甚远。此外,动态量子蒙特卡罗结果与 CCSD(T) 数值相差数十 kcal/mol。与一系列流行的密度泛函产生的 DFT 结果的比较显示了预期的结果散射,并展示了将 DFT 应用到自旋态能量的难度。










































 京公网安备 11010802027423号
京公网安备 11010802027423号