Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structure and orientation effects in the coalescence of Au clusters
Nanoscale ( IF 5.8 ) Pub Date : 2020/03/19 , DOI: 10.1039/c9nr10163b Diana Nelli 1, 2, 3, 4 , Giulia Rossi 1, 2, 3, 4 , Zhiwei Wang 5, 6, 7, 8, 9 , Richard E. Palmer 5, 6, 7, 8 , Riccardo Ferrando 4, 10, 11
Nanoscale ( IF 5.8 ) Pub Date : 2020/03/19 , DOI: 10.1039/c9nr10163b Diana Nelli 1, 2, 3, 4 , Giulia Rossi 1, 2, 3, 4 , Zhiwei Wang 5, 6, 7, 8, 9 , Richard E. Palmer 5, 6, 7, 8 , Riccardo Ferrando 4, 10, 11
Affiliation
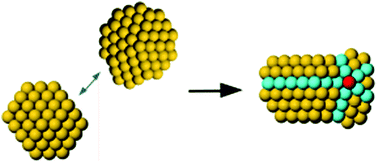
|
The atomic configuration of nanoparticles is key to their properties, in a way that resembles the structure–function correlation of biomolecules, and depends on the growth mechanism. Coalescence is often an important step in the growth of nanoparticles, both in liquid and in the gas phase. In coalescence, two preformed clusters collide and merge to form a larger aggregate, whose shape evolves from an initial configuration, which is strongly out of equilibrium, towards more compact structures. Here the coalescence in the gas phase of gold clusters of size ∼2 nm is simulated by the Molecular Dynamics (MD) method. Our simulations reveal a persistent influence of the structure and relative orientation of the initial colliding clusters on the final coalesced aggregates, even after more than 1 μs at temperature T = 500 K. This result is interpreted in terms of a special type of kinetic trapping, in which the choice of the structural motif takes place in the initial stages of the coalescence process. In the subsequent evolution, the coalescing aggregate may equilibrate within that motif, while transformations between different motifs are not observed, so that inter-motif equilibration is not achieved. Inter-motif equilibration is only achieved at 550 K and above. The simulations also show that aggregate reshaping occurs by a variety of specific transformation pathways, which often involve the concerted displacements of many atoms.
中文翻译:

金团簇聚结中的结构和取向效应
纳米粒子的原子构型是其性能的关键,其方式类似于生物分子的结构-功能相关性,并取决于生长机理。聚结通常是液相和气相中纳米颗粒生长的重要步骤。在聚结中,两个预形成的团簇碰撞并合并形成一个较大的聚集体,其形状从强烈不平衡的初始构型演变为更紧凑的结构。这里,通过分子动力学(MD)方法模拟了大小约为2 nm的金团簇在气相中的聚结。我们的模拟揭示了即使在温度T超过1μs之后,初始碰撞簇的结构和相对取向对最终聚结团聚体的持续影响。= 500K。此结果是根据一种特殊的动力学陷阱来解释的,其中结构基序的选择发生在合并过程的初始阶段。在随后的演变中,聚结聚集体可能在该基序内达到平衡,而未观察到不同基序之间的转化,因此无法实现基序间的平衡。图案间平衡仅在550 K及以上时实现。模拟还表明,聚集体重塑通过各种特定的转化途径发生,这些途径通常涉及许多原子的协调位移。
更新日期:2020-04-09
中文翻译:
金团簇聚结中的结构和取向效应
纳米粒子的原子构型是其性能的关键,其方式类似于生物分子的结构-功能相关性,并取决于生长机理。聚结通常是液相和气相中纳米颗粒生长的重要步骤。在聚结中,两个预形成的团簇碰撞并合并形成一个较大的聚集体,其形状从强烈不平衡的初始构型演变为更紧凑的结构。这里,通过分子动力学(MD)方法模拟了大小约为2 nm的金团簇在气相中的聚结。我们的模拟揭示了即使在温度T超过1μs之后,初始碰撞簇的结构和相对取向对最终聚结团聚体的持续影响。= 500K。此结果是根据一种特殊的动力学陷阱来解释的,其中结构基序的选择发生在合并过程的初始阶段。在随后的演变中,聚结聚集体可能在该基序内达到平衡,而未观察到不同基序之间的转化,因此无法实现基序间的平衡。图案间平衡仅在550 K及以上时实现。模拟还表明,聚集体重塑通过各种特定的转化途径发生,这些途径通常涉及许多原子的协调位移。








































 京公网安备 11010802027423号
京公网安备 11010802027423号