当前位置:
X-MOL 学术
›
Bioorgan. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Development and biological investigations of hypoxia-sensitive prodrugs of the tyrosine kinase inhibitor crizotinib.
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2020-03-20 , DOI: 10.1016/j.bioorg.2020.103778 Bjoern Bielec 1 , Hemma Schueffl 2 , Alessio Terenzi 3 , Walter Berger 4 , Petra Heffeter 4 , Bernhard K Keppler 5 , Christian R Kowol 5
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2020-03-20 , DOI: 10.1016/j.bioorg.2020.103778 Bjoern Bielec 1 , Hemma Schueffl 2 , Alessio Terenzi 3 , Walter Berger 4 , Petra Heffeter 4 , Bernhard K Keppler 5 , Christian R Kowol 5
Affiliation
|
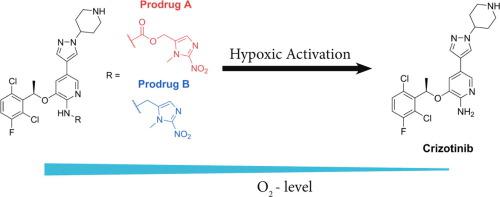
Despite the huge success of tyrosine kinase inhibitors as anticancer agents, severe side effects are a major problem. In order to overcome this drawback, the first hypoxia-activatable 2-nitroimidazole-based prodrugs of the clinically approved ALK and c-MET inhibitor crizotinib were developed. The 2-aminopyridine functionality of crizotinib (essential for target kinase binding) was considered as ideal position for prodrug derivatization. Consequently, two different prodrugs were synthesized with the nitroimidazole unit attached to crizotinib either via carbamoylation (A) or alkylation (B) of the 2-aminopyridine moiety. The successful prodrug design could be proven by docking studies and a dramatically reduced ALK and c-MET kinase-inhibitory potential. Furthermore, the prodrugs showed high stability in serum and release of crizotinib in an enzymatic nitroreductase-based cleavage assay was observed for prodrug A. The in vitro activity of both prodrugs was investigated against ALK- and c-MET-dependent or -overexpressing cells, revealing a distinct hypoxia-dependent activation for prodrug A. Finally, inhibition of c-MET phosphorylation and cell proliferation could also be proven in vivo. In summary of the theoretical, chemical and biological studies, prodrug derivatization of the 2-aminopyridine position can be considered as a promising strategy to reduce the side effects and improve the anticancer activity of crizotinib.
中文翻译:

酪氨酸激酶抑制剂克唑替尼的缺氧敏感性前药的开发和生物学研究。
尽管酪氨酸激酶抑制剂作为抗癌剂取得了巨大成功,但严重的副作用是一个主要问题。为了克服这个缺点,临床批准的 ALK 和 c-MET 抑制剂克唑替尼的第一个缺氧可激活的基于 2-硝基咪唑的前药被开发出来。克唑替尼的 2-氨基吡啶官能团(对靶激酶结合必不可少)被认为是前药衍生化的理想位置。因此,合成了两种不同的前药,其中硝基咪唑单元通过 2-氨基吡啶部分的氨基甲酰化 (A) 或烷基化 (B) 连接到克唑替尼。成功的前药设计可以通过对接研究和显着降低的 ALK 和 c-MET 激酶抑制潜力来证明。此外,前药在血清中表现出高稳定性,并且在基于酶促硝基还原酶的裂解试验中观察到克唑替尼的释放对前药 A 而言。前药 A 具有明显的缺氧依赖性激活。最后,还可以在体内证明对 c-MET 磷酸化和细胞增殖的抑制。总结理论、化学和生物学研究,2-氨基吡啶位置的前药衍生化可以被认为是减少副作用和提高克唑替尼抗癌活性的有前途的策略。揭示了前药 A 的明显缺氧依赖性激活。最后,还可以在体内证明对 c-MET 磷酸化和细胞增殖的抑制。总结理论、化学和生物学研究,2-氨基吡啶位置的前药衍生化可以被认为是减少副作用和提高克唑替尼抗癌活性的有前途的策略。揭示了前药 A 的明显缺氧依赖性激活。最后,还可以在体内证明对 c-MET 磷酸化和细胞增殖的抑制。总结理论、化学和生物学研究,2-氨基吡啶位置的前药衍生化可以被认为是减少副作用和提高克唑替尼抗癌活性的有前途的策略。
更新日期:2020-04-20
中文翻译:
酪氨酸激酶抑制剂克唑替尼的缺氧敏感性前药的开发和生物学研究。
尽管酪氨酸激酶抑制剂作为抗癌剂取得了巨大成功,但严重的副作用是一个主要问题。为了克服这个缺点,临床批准的 ALK 和 c-MET 抑制剂克唑替尼的第一个缺氧可激活的基于 2-硝基咪唑的前药被开发出来。克唑替尼的 2-氨基吡啶官能团(对靶激酶结合必不可少)被认为是前药衍生化的理想位置。因此,合成了两种不同的前药,其中硝基咪唑单元通过 2-氨基吡啶部分的氨基甲酰化 (A) 或烷基化 (B) 连接到克唑替尼。成功的前药设计可以通过对接研究和显着降低的 ALK 和 c-MET 激酶抑制潜力来证明。此外,前药在血清中表现出高稳定性,并且在基于酶促硝基还原酶的裂解试验中观察到克唑替尼的释放对前药 A 而言。前药 A 具有明显的缺氧依赖性激活。最后,还可以在体内证明对 c-MET 磷酸化和细胞增殖的抑制。总结理论、化学和生物学研究,2-氨基吡啶位置的前药衍生化可以被认为是减少副作用和提高克唑替尼抗癌活性的有前途的策略。揭示了前药 A 的明显缺氧依赖性激活。最后,还可以在体内证明对 c-MET 磷酸化和细胞增殖的抑制。总结理论、化学和生物学研究,2-氨基吡啶位置的前药衍生化可以被认为是减少副作用和提高克唑替尼抗癌活性的有前途的策略。揭示了前药 A 的明显缺氧依赖性激活。最后,还可以在体内证明对 c-MET 磷酸化和细胞增殖的抑制。总结理论、化学和生物学研究,2-氨基吡啶位置的前药衍生化可以被认为是减少副作用和提高克唑替尼抗癌活性的有前途的策略。










































 京公网安备 11010802027423号
京公网安备 11010802027423号