当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Kinetic Selection and Relaxation of the Intrinsically Disordered Region of a Protein upon Binding
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-19 , DOI: 10.1021/acs.jctc.9b01203 Duy Phuoc Tran 1 , Akio Kitao 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-19 , DOI: 10.1021/acs.jctc.9b01203 Duy Phuoc Tran 1 , Akio Kitao 1
Affiliation
|
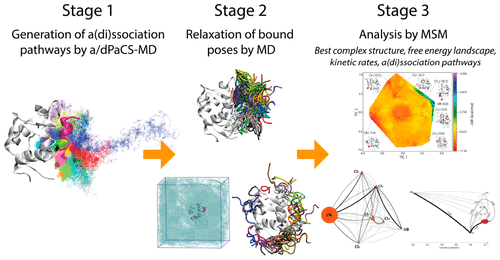
Here, we investigate the association and dissociation mechanisms of a typical intrinsically disordered region (IDR), transcriptional activation subdomain of tumor suppressor protein p53 (TAD-p53), with murine double-minute clone 2 protein (MDM2). Using a combination of cycles of association and dissociation parallel cascade molecular dynamics, multiple standard molecular dynamics (MD), and the Markov state model, we were successful in obtaining the lowest free energy structure of the MDM2/TAD-p53 complex as the structure closest to the crystal structure without prior knowledge of the crystal structure. This method also reproduced the experimentally measured standard binding free energy, and the association and dissociation rate constants, requiring only an accumulated MD simulation cost of 11.675 μs even though that actual dissociation occurs on the order of seconds. We identified few complex intermediates with similar free energies; yet TAD-p53 first binds MDM2 as the second lowest free energy intermediate kinetically with >90% of the flux, adopting a conformation similar to that of one of these few intermediates in its monomeric state. Even though the mechanism of the first step has a conformational-selection-type aspect, the second step shows induced-fit-like features and occurs as concomitant dehydration of the interface, side-chain π–π stacking, and main-chain hydrogen-bond formation to complete binding as an α-helix. In addition, dehydration is a key process for the final relaxation process around the complex interface. These results demonstrate that TAD-p53 kinetically selects its initial binding form and then relaxes to complete the binding.
中文翻译:

结合时蛋白质固有紊乱区域的动力学选择和松弛
在这里,我们调查了典型的内在无序区(IDR),肿瘤抑制蛋白p53(TAD-p53)的转录激活亚结构域与鼠类双重克隆2蛋白(MDM2)的缔合和解离机制。通过结合和解离循环的并行级联分子动力学,多个标准分子动力学(MD)和马尔可夫状态模型的组合,我们成功地获得了MDM2 / TAD-p53配合物的最低自由能结构作为最接近的结构没有晶体结构的先验知识的晶体结构。此方法还重现了实验测量的标准结合自由能以及缔合和解离速率常数,仅需要累积的MD模拟成本为11。即使实际的解离发生在几秒的数量级,也为675μs。我们发现很少有具有相似自由能的复杂中间体。然而,TAD-p53首先以大于90%的通量将MDM2作为第二低的自由能中间体进行动力学结合,其构象类似于上述几种处于单体状态的中间体之一。即使第一步的机制具有构象选择型方面,第二步仍显示出诱导拟合的特征,并且发生在界面脱水,侧链π-π堆积和主链氢-键形成完整结合的α-螺旋。此外,脱水是围绕复杂界面进行最终松弛过程的关键过程。
更新日期:2020-04-24
中文翻译:
结合时蛋白质固有紊乱区域的动力学选择和松弛
在这里,我们调查了典型的内在无序区(IDR),肿瘤抑制蛋白p53(TAD-p53)的转录激活亚结构域与鼠类双重克隆2蛋白(MDM2)的缔合和解离机制。通过结合和解离循环的并行级联分子动力学,多个标准分子动力学(MD)和马尔可夫状态模型的组合,我们成功地获得了MDM2 / TAD-p53配合物的最低自由能结构作为最接近的结构没有晶体结构的先验知识的晶体结构。此方法还重现了实验测量的标准结合自由能以及缔合和解离速率常数,仅需要累积的MD模拟成本为11。即使实际的解离发生在几秒的数量级,也为675μs。我们发现很少有具有相似自由能的复杂中间体。然而,TAD-p53首先以大于90%的通量将MDM2作为第二低的自由能中间体进行动力学结合,其构象类似于上述几种处于单体状态的中间体之一。即使第一步的机制具有构象选择型方面,第二步仍显示出诱导拟合的特征,并且发生在界面脱水,侧链π-π堆积和主链氢-键形成完整结合的α-螺旋。此外,脱水是围绕复杂界面进行最终松弛过程的关键过程。









































 京公网安备 11010802027423号
京公网安备 11010802027423号