当前位置:
X-MOL 学术
›
ChemPhysChem
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The Effect of Phenyl Substitutions on Microstructures and Dynamics of Tetraalkylphosphonium Bis(trifluoro- methylsulfonyl)imide Ionic Liquids.
ChemPhysChem ( IF 2.3 ) Pub Date : 2020-04-21 , DOI: 10.1002/cphc.201901206 Yong-Lei Wang 1 , Bin Li 2 , Aatto Laaksonen 1, 3 , Jiayin Yuan 1
ChemPhysChem ( IF 2.3 ) Pub Date : 2020-04-21 , DOI: 10.1002/cphc.201901206 Yong-Lei Wang 1 , Bin Li 2 , Aatto Laaksonen 1, 3 , Jiayin Yuan 1
Affiliation
|
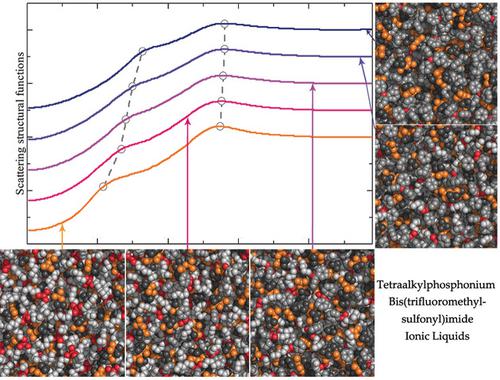
Extensive atomistic simulations demonstrated that a gradual substitution of hexyl chains with phenyl groups in tetraalkylphosphonium cations results in remarkable changes in hydrogen bonding interactions, liquid structures and scattering structural functions, and rotational dynamics of hexyl chains and phenyl groups in tetraalkylphosphonium bis(trifluoromethylsulfonyl)imide ionic liquids. Hydrogen donor sites in hexyl chains present competitive characteristics with those in phenyl groups in coordinating anions, as well as their continuous and intermittent hydrogen bonding dynamics. Cation‐cation and anion‐anion spatial correlations show concomitant shift to short distances with decreased peak intensities with variations of cation structures, whereas cation‐anion correlations have a distinct shift to large radial distances due to decreased associations of anions with neighboring cations. These microstructural changes are qualitatively manifested in shifts of prominent peaks for prevalent charge alternations and adjacency correlations between ion species in scattering structural functions. Meanwhile, rotational dynamics of hexyl chains speed up, which, in turn, slow down rotations of phenyl groups, whereas anions exhibit imperceptible changes in their rotational dynamics. These computational results are intrinsically correlated with conformational flexibilities, molecular sizes, and steric hindrance effects of phenyl groups in comparison with hexyl chains, and constrained distributions of anions around cations in heterogeneous ionic environments.
中文翻译:

苯基取代对四烷基phosph双(三氟-甲基磺酰基)亚胺离子液体的微观结构和动力学的影响。
广泛的原子模拟表明,四烷基phosph阳离子中的苯基逐渐取代己基链会导致氢键相互作用,液体结构和散射结构功能的显着变化,以及四烷基phosph双(三氟甲基磺酰基)酰亚胺离子中己基链和苯基的旋转动力学。液体。在配位阴离子中,己基链中的氢供体位点与苯基中的氢供体位点表现出竞争特征,以及它们的连续和间歇性氢键动力学。阳离子-阳离子和阴离子-阴离子的空间相关性表明,伴随阳离子结构的变化,伴随着峰强度的减小,伴随着向短距离的转移,而由于阴离子与相邻阳离子的缔合减少,阳离子-阴离子的相关性向径向距离的变化明显。这些微观结构的变化定性地表现在显着峰的位移上,这些峰用于普遍的电荷交替以及散射结构功能中离子种类之间的邻接关系。同时,己基链的旋转动力学加快,这反过来又减慢了苯基的旋转,而阴离子的旋转动力学却发生了不可察觉的变化。与己基链相比,这些计算结果与苯基的构象柔韧性,分子大小和空间位阻效应具有内在联系,并且在异质离子环境中阳离子周围的阴离子分布受限。这些微观结构的变化定性地表现在显着峰的位移上,这些峰用于普遍的电荷交替以及散射结构功能中离子种类之间的邻接关系。同时,己基链的旋转动力学加快,这反过来又减慢了苯基的旋转,而阴离子的旋转动力学却发生了不可察觉的变化。这些计算结果与苯基的结构柔韧性,分子大小和与己基链相比的位阻效应具有内在联系,并且在非离子环境中阳离子周围的阴离子分布受到约束。这些微观结构的变化定性地表现在显着峰的位移上,这些峰用于普遍的电荷交替以及散射结构功能中离子种类之间的邻接关系。同时,己基链的旋转动力学加快,这反过来又减慢了苯基的旋转,而阴离子的旋转动力学却发生了不可察觉的变化。与己基链相比,这些计算结果与苯基的构象柔韧性,分子大小和空间位阻效应具有内在联系,并且在异质离子环境中阳离子周围的阴离子分布受限。
更新日期:2020-04-21
中文翻译:
苯基取代对四烷基phosph双(三氟-甲基磺酰基)亚胺离子液体的微观结构和动力学的影响。
广泛的原子模拟表明,四烷基phosph阳离子中的苯基逐渐取代己基链会导致氢键相互作用,液体结构和散射结构功能的显着变化,以及四烷基phosph双(三氟甲基磺酰基)酰亚胺离子中己基链和苯基的旋转动力学。液体。在配位阴离子中,己基链中的氢供体位点与苯基中的氢供体位点表现出竞争特征,以及它们的连续和间歇性氢键动力学。阳离子-阳离子和阴离子-阴离子的空间相关性表明,伴随阳离子结构的变化,伴随着峰强度的减小,伴随着向短距离的转移,而由于阴离子与相邻阳离子的缔合减少,阳离子-阴离子的相关性向径向距离的变化明显。这些微观结构的变化定性地表现在显着峰的位移上,这些峰用于普遍的电荷交替以及散射结构功能中离子种类之间的邻接关系。同时,己基链的旋转动力学加快,这反过来又减慢了苯基的旋转,而阴离子的旋转动力学却发生了不可察觉的变化。与己基链相比,这些计算结果与苯基的构象柔韧性,分子大小和空间位阻效应具有内在联系,并且在异质离子环境中阳离子周围的阴离子分布受限。这些微观结构的变化定性地表现在显着峰的位移上,这些峰用于普遍的电荷交替以及散射结构功能中离子种类之间的邻接关系。同时,己基链的旋转动力学加快,这反过来又减慢了苯基的旋转,而阴离子的旋转动力学却发生了不可察觉的变化。这些计算结果与苯基的结构柔韧性,分子大小和与己基链相比的位阻效应具有内在联系,并且在非离子环境中阳离子周围的阴离子分布受到约束。这些微观结构的变化定性地表现在显着峰的位移上,这些峰用于普遍的电荷交替以及散射结构功能中离子种类之间的邻接关系。同时,己基链的旋转动力学加快,这反过来又减慢了苯基的旋转,而阴离子的旋转动力学却发生了不可察觉的变化。与己基链相比,这些计算结果与苯基的构象柔韧性,分子大小和空间位阻效应具有内在联系,并且在异质离子环境中阳离子周围的阴离子分布受限。









































 京公网安备 11010802027423号
京公网安备 11010802027423号