当前位置:
X-MOL 学术
›
Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Density Functional Theory study of the SiF molecule adsorption and decomposition on p(2 × 2) reconstructed Si(001) surface
Surface Science ( IF 2.1 ) Pub Date : 2020-07-01 , DOI: 10.1016/j.susc.2020.121602 L. Bouamama , A. Lounis , A. Mokrani , A. Ziane , S. Bouarab , A. Rhallabi
Surface Science ( IF 2.1 ) Pub Date : 2020-07-01 , DOI: 10.1016/j.susc.2020.121602 L. Bouamama , A. Lounis , A. Mokrani , A. Ziane , S. Bouarab , A. Rhallabi
|
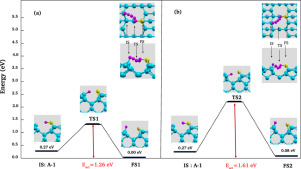
Abstract The adsorption of the F and Si atoms as well as the SiF molecule on the Si(001)-p(2×2) surface is studied using first-principle calculations. Our results show that the fluorine atom saturates the dangling bond of the surface while the silicon adatom forms two bonds with the surface atoms. For the adsorption of the SiF molecule, we obtained one dissociative state which is the most stable one and seven non-dissociative states. The minimum energy paths for F diffusion and SiF decomposition on the silicon surface are explored using the nudged elastic band (NEB) method. Our study reveals that it is energetically favorable for the fluorine atom to diffuse from the SiF molecule to a surface site than the diffusion from surface site to another one.
中文翻译:

在 p(2 × 2) 重构 Si(001) 表面上 SiF 分子吸附和分解的密度泛函理论研究
摘要 使用第一性原理计算研究了 F 和 Si 原子以及 SiF 分子在 Si(001)-p(2×2) 表面上的吸附。我们的结果表明,氟原子使表面的悬空键饱和,而硅原子与表面原子形成两个键。对于 SiF 分子的吸附,我们获得了一种解离状态,即最稳定的一种和七种非解离状态。使用轻推弹性带 (NEB) 方法探索了硅表面上 F 扩散和 SiF 分解的最小能量路径。我们的研究表明,氟原子从 SiF 分子扩散到表面位点比从表面位点扩散到另一个位点在能量上更有利。
更新日期:2020-07-01
中文翻译:
在 p(2 × 2) 重构 Si(001) 表面上 SiF 分子吸附和分解的密度泛函理论研究
摘要 使用第一性原理计算研究了 F 和 Si 原子以及 SiF 分子在 Si(001)-p(2×2) 表面上的吸附。我们的结果表明,氟原子使表面的悬空键饱和,而硅原子与表面原子形成两个键。对于 SiF 分子的吸附,我们获得了一种解离状态,即最稳定的一种和七种非解离状态。使用轻推弹性带 (NEB) 方法探索了硅表面上 F 扩散和 SiF 分解的最小能量路径。我们的研究表明,氟原子从 SiF 分子扩散到表面位点比从表面位点扩散到另一个位点在能量上更有利。










































 京公网安备 11010802027423号
京公网安备 11010802027423号