当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Performance of Mixed Quantum-Classical Approaches on Modeling the Crossover from Hopping to Bandlike Charge Transport in Organic Semiconductors
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jctc.9b01271 Weiwei Xie 1 , Daniel Holub 1 , Tomáš Kubař 1 , Marcus Elstner 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-03-16 , DOI: 10.1021/acs.jctc.9b01271 Weiwei Xie 1 , Daniel Holub 1 , Tomáš Kubař 1 , Marcus Elstner 1, 2
Affiliation
|
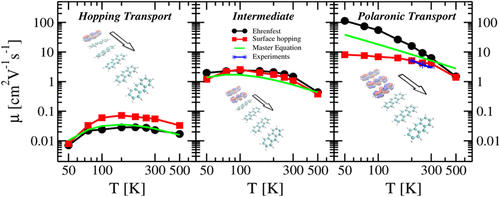
In the present study, several mixed quantum-classical (MQC) methods are applied to on-the-fly nonadiabatic molecular dynamics simulations of hole transport in molecular organic semiconductors (OSCs). The tested MQC methods contain the mean-field Ehrenfest (MFE), trajectory surface hopping (TSH) approaches based on Tully’s fewest switches surface hopping (FSSH) and the global flux surface hopping (GFSH), the latter in the diabatic/adiabatic representation, and a Landau–Zener type trajectory surface hopping (LZSH). We also tested several correction schemes which were proposed to identify trivial crossings and to remove unphysical long-range charge transfers due to decoherence corrections. In addition, several cost-effective approaches for the nuclear velocity adjustment after an energy-allowed/energy-forbidden hop are investigated with respect to detailed balance and internal consistency conditions. To model a broad spectrum of OSCs with different charge transport characteristics, we derived from the anthracene structural model the construction of two additional models by uniformly scaling down the electronic couplings by the factors of 0.1 and 0.5. Anthracene shows a bandlike charge transport mechanism, characterized by slightly delocalized charge carriers ‘diffusing’ through the crystal. For smaller couplings, the mechanism changes to a hopping type, characteristically differing in the charge delocalization and temperature dependence. The MFE and corrected adiabatic TSH approaches are able to quantitatively reproduce the expected behavior, while the diabatic LZSH method fails for large couplings, as do approaches which are based on the hopping of localized charge between neighboring sites. Moreover, we find that while the hole mobility of the anthracene crystal simulated using the celebrated Marcus theory is in good agreement with the experimental value, its agreement has to be regarded as an accident due to the overestimation of the prefactor in the Marcus rate equation.
中文翻译:

混合量子经典方法在有机半导体中从跳跃跃迁到带状电荷传输的过程中的性能
在本研究中,几种混合的量子经典方法(MQC)被应用于动态非绝热分子动力学模拟的分子有机半导体(OSC)中的空穴传输。经过测试的MQC方法包含均场Ehrenfest(MFE),基于Tully最少的开关表面跳变(FSSH)和全局通量表面跳变(GFSH)的轨迹表面跳变(TSH)方法,后者以非绝热/绝热表示形式,以及Landau-Zener型轨迹表面跳变(LZSH)。我们还测试了几种校正方案,这些方案被提议用来识别微不足道的交叉并消除由于去相干校正而引起的非物理性远距离电荷转移。此外,针对详细的平衡和内部一致性条件,研究了几种在允许能量/禁止能量跃变之后进行核速度调整的经济有效方法。为了建模具有不同电荷传输特性的多种OSC,我们从蒽结构模型中获得了两个附加模型的构建,即通过将电子耦合均匀缩小0.1和0.5倍。蒽显示出带状电荷传输机制,其特征是略微离域的载流子“扩散”通过晶体。对于较小的耦合,该机制将变为跳变类型,其特征在于电荷离域和温度依赖性不同。MFE和校正的绝热TSH方法能够定量再现预期的行为,而绝热的LZSH方法无法进行大的耦合,就像基于相邻站点之间局部电荷跳跃的方法一样。此外,我们发现,尽管使用著名的马库斯理论模拟的蒽晶体的空穴迁移率与实验值非常吻合,但由于对马库斯速率方程中前因子的高估,其一致性被认为是偶然的。
更新日期:2020-04-24
中文翻译:
混合量子经典方法在有机半导体中从跳跃跃迁到带状电荷传输的过程中的性能
在本研究中,几种混合的量子经典方法(MQC)被应用于动态非绝热分子动力学模拟的分子有机半导体(OSC)中的空穴传输。经过测试的MQC方法包含均场Ehrenfest(MFE),基于Tully最少的开关表面跳变(FSSH)和全局通量表面跳变(GFSH)的轨迹表面跳变(TSH)方法,后者以非绝热/绝热表示形式,以及Landau-Zener型轨迹表面跳变(LZSH)。我们还测试了几种校正方案,这些方案被提议用来识别微不足道的交叉并消除由于去相干校正而引起的非物理性远距离电荷转移。此外,针对详细的平衡和内部一致性条件,研究了几种在允许能量/禁止能量跃变之后进行核速度调整的经济有效方法。为了建模具有不同电荷传输特性的多种OSC,我们从蒽结构模型中获得了两个附加模型的构建,即通过将电子耦合均匀缩小0.1和0.5倍。蒽显示出带状电荷传输机制,其特征是略微离域的载流子“扩散”通过晶体。对于较小的耦合,该机制将变为跳变类型,其特征在于电荷离域和温度依赖性不同。MFE和校正的绝热TSH方法能够定量再现预期的行为,而绝热的LZSH方法无法进行大的耦合,就像基于相邻站点之间局部电荷跳跃的方法一样。此外,我们发现,尽管使用著名的马库斯理论模拟的蒽晶体的空穴迁移率与实验值非常吻合,但由于对马库斯速率方程中前因子的高估,其一致性被认为是偶然的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号